A protein called RepID is key to regulating DNA replication and preventing cancer when the process goes awry.
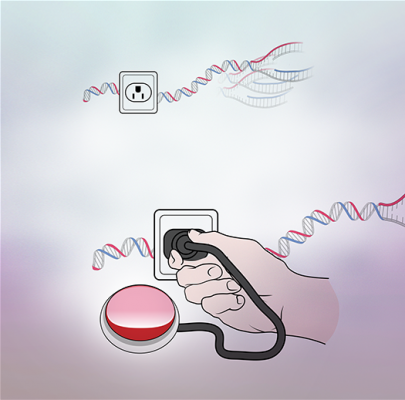
The electrical socket in this image represents specific sites on chromatin (replication origins) that can bind a protein (RepID, depicted as a hand) that recruits a regulator complex (CRL4, a red button). Without RepID (top panel), the regulator complex is absent, and replication might start at the same chromosome sites more than once (right). With RepID (bottom panel), the controller CRL4 plugs into chromatin and prevents replication from starting more than once at each genomic location, ensuring that the entire genome replicates exactly once before each cell division.
Credit: Sang-Min Jang, CCR, NCI, NIH and Erina He, Medical Arts, NIH
Each time a cell divides, it must first duplicate its genetic material in a process called DNA replication. Because defects in this process can cause mutations that eventually lead to cancer, understanding the details of how replication works could point to possible cancer treatments. Now, CCR scientists have added to this understanding by describing novel aspects of DNA replication that involve a protein called RepID.
Cells have a number of molecular checkpoints in place to prevent DNA replication from going awry, including some to ensure that cells replicate their DNA only once before each division. Occasionally, these checkpoints get disrupted. As a result, cells can become cancerous through a variety of mechanisms, such as increased copies of genes that drive cancer growth or through uncontrollable cell division, which is a hallmark of cancer.
DNA replication errors, especially those occurring at regions that are hard to replicate, called fragile sites, can cause breaks in DNA. This can lead to cancer, primarily by making it more likely that fragments of chromosomes rearrange themselves, activating genes that lead to uncontrollable cell division.
A team led by Mirit Aladjem, Ph.D., Senior Investigator in CCR’s Developmental Therapeutics Branch, and Sang-Min Jang, Ph.D., a postdoctoral researcher in Aladjem’s lab, reported in Nature Communications that the RepID protein sets off a chain reaction of protein signaling and recruitment to prevent DNA replication from getting out of control. They showed that RepID recruits the enzyme CRL4 to chromatin before DNA replication begins. Once CRL4 is bound to chromatin, it causes a protein called CDT1 to break down, keeping it from kick-starting replication more than once each time cells divide. If CRL4 is not working properly, though, a buildup of CDT1 can occur, allowing DNA replication to proceed unchecked, resulting in excess replication of segments of the genome.

Oftentimes, portions of DNA replication checkpoints are disrupted in cancer cells. A number of drugs under investigation inhibit the remaining functional checkpoints in these cells, triggering excessive DNA replication. This places an excessive burden on the cancer cells, leading to improper separation of their chromosomes when they divide. The ultimate result is programmed cell death, or apoptosis. In other words, malfunctioning DNA replication checkpoints may actually help kill cancer cells because they render them more sensitive to this class of drugs.
Aladjem and her team tested one such drug, and they found that the drug triggered severe excess DNA replication in cells depleted of RepID. As a result, a greater proportion of RepID-depleted cells than RepID-containing cells underwent apoptosis. These results point to RepID expression levels as a way to gauge cancer cells’ sensitivity to replication inhibitors.
Aladjem notes that her team’s research explains the underlying mechanisms cells use to prevent DNA from replicating more than once each cell cycle. These basic findings will improve our understanding of not only how normal cells avoid becoming cancerous, but also how certain drugs can exploit excessive DNA replication to kill cancer cells.