Activating the cellular stress response puts the brakes on overactive signaling by one of cancer’s most notorious growth promoters.
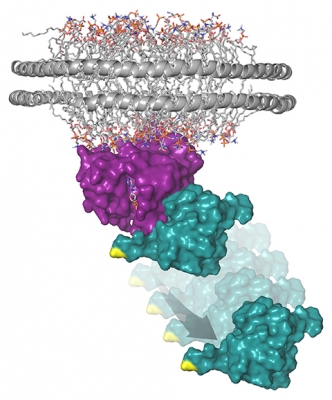
This model represents the structure of the Ras protein (purple) attached to a membrane-like nanodisc with the Raf-RBD (teal) protein domain bound to RAS. In response to cellular stresses, like rigosertib treatment, JNK becomes activated and phosphorylates (or modifies) Raf on a specific site (shown in yellow) that disrupts binding between RAS and Raf.
Credit: Deborah Morrison, CCR, NCI, NIH
New research from Deborah Morrison, Ph.D., Chief of the Laboratory of Cell and Developmental Signaling, shows that conditions of cellular stress can halt signaling from the growth-promoting RAS pathway, which drives the development of many cancers and has been notoriously difficult to stop.
More than a third of human cancers, including about 95 percent of pancreatic cancers and 45 percent of colorectal cancers, carry mutations in RAS genes. In healthy cells, the proteins encoded by these genes help relay growth-triggering signals from outside a cell to the gene regulators that spur appropriate cell proliferation. Mutations can lock Ras proteins into a position where they send their growth-promoting signals all the time, driving tumor development.
Researchers have tried for decades to develop drugs that stop tumor growth by blocking dysfunctional RAS signaling but have found the protein to be a particularly difficult target. Because creating effective new treatments for RAS-driven cancers would have such an enormous impact, in 2013 NCI established a major initiative to bring a wide range of resources and expertise to bear on this problem. Working together under the RAS Initiative, researchers in government, industry and academia are learning new details of how the RAS pathway works and laying the groundwork for drug development.
Morrison, a collaborator in the initiative, has long been teasing out the precise mechanisms that enzymes in the RAS pathway use to relay signals that regulate cell growth. An investigational drug called rigosertib, which blocks the growth of cancerous cells in the lab, caught her team’s attention because it was reported to interfere with the RAS pathway, possibly by preventing RAS from interacting with its signaling partners.
When Morrison and her colleagues took a close look at the drug’s effects, however, they found, as they reported in Molecular Cell, that its effects on RAS signaling are not so direct. Rather than interacting with proteins in the RAS pathway, rigosertib causes cellular changes that trigger a cell’s stress response. The stress-response pathway, in turn, interrupts RAS signaling to put the brakes on cell growth.
The interaction between the two pathways appears to be a signaling checkpoint that ensures that cells suspend their growth while they assess and respond to stressful conditions, Morrison says. In detailed biochemical experiments, she and RAS Initiative collaborators at the Frederick National Laboratory for Cancer Research teased out exactly how a stress-response pathway, called the JNK pathway, switches off specific signaling molecules in the RAS pathway, effectively blocking RAS-mediated growth signals.
Rigosertib is currently being evaluated in clinical trials as a treatment for myelodysplastic syndromes, in which immature blood cells in the bone marrow fail to mature. Morrison’s team’s findings could help researchers identify new ways to use the compound. In particular, she says, the new understanding of rigosertib’s activity suggests that it might be useful in preventing or slowing the development of drug resistance caused by increased RAS signaling when used in combination with other cancer therapies.
Reference
Ritt DA, et al. Mol Cell. 2016 Dec 1; 64(5):875-887.


