
Alex Compton, Ph.D.
- Center for Cancer Research
- National Cancer Institute
- Building 535, Room 307
- Frederick, MD 21702-1201
- 301-846-7144
- 301-846-6013
- alex.compton@nih.gov
RESEARCH SUMMARY
Work in the Antiviral Immunity and Resistance Section, led by Alex Compton, Ph.D., is guided by integrative approaches combining experimental virology, cell biology, and evolutionary biology to reveal dynamic host-virus interactions important for public health. A focus is placed on mechanisms of host protection mediated by the cell-intrinsic innate immune response, as well as the strategies employed by HIV and emerging viruses to evade or overcome these immune barriers.
Areas of Expertise
Alex Compton, Ph.D.
Research
Improving Human Antiviral Immunity, One Cell at a Time
The clinical outcome of viral infection, the difference between survival and death of the host, rests delicately on events occurring at the molecular level of individual cells. The "cell-intrinsic" arm of innate immunity prevents virus replication in host cells by detecting virus invasion and interfering with the viral life cycle. As such, cell-intrinsic immune factors, also known as host restriction factors, impose the earliest-acting barriers to invading pathogens. Research in this field, which relies increasingly on interdisciplinary approaches and bioinformatics, has demonstrated that the survival of single cells equates strongly with survival of the organism, and even of the population (or species) to which it belongs. Since its inception in 2017, the Compton lab focuses on mechanisms of cell-intrinsic immunity and the strategies employed by HIV and emerging viruses to evade or overcome these immune barriers.
By combining relevant experimental systems in virology with perspectives in cell biology and evolutionary biology, we aim to better understand the factors governing virus entry into cells. We employ a “cross-species” approach, in which diverse viruses are paired with host cells of diverse species in order to reveal cell-intrinsic barriers that limit virus infection. Cellular membranes, composed of proteins and lipids, are the first line of defense against infection. Residents of this critical threshold include cellular transmembrane proteins that remodel membrane vesicles or redirect their trafficking in order to inhibit the viral entry process. In addition to protecting cells from infection, cellular membrane components also impact the structure and infectivity of nascent virions produced from infected cells. Overall, cell-intrinsic immunity acting on membranes performs dual antiviral functions by 1) preventing virus infection of individual cells, and 2) limiting the spread of virus between cells.
Current projects in the lab revolve around the following themes:
1. Mechanisms of virus entry into cells and evasion of cell-intrinsic immunity
2. Enhancement of virus delivery for gene editing in human cells and in vivo
3. Signals regulating the intrinsic antiviral state: stress, metabolism, differentiation, and activation
Biography

Alex Compton, Ph.D.
Dr. Alex Compton received his Ph.D. in Molecular and Cellular Biology from the University of Washington in 2012. As a doctoral student in the laboratory of Dr. Michael Emerman (Fred Hutchinson Cancer Research Center), he received an NSF Graduate Research Fellowship and characterized the coevolution of primate lentiviruses with their hosts. Later, Dr. Compton was the recipient of a competitive postdoctoral fellowship from the Louis Pasteur Foundation to support his work at Institut Pasteur in Paris, France. In the lab of Dr. Olivier Schwartz, he provided important insight into the complex ways in which mammalian cells have evolved to block virus entry into cells. In 2017, Dr. Compton joined the HIV Dynamics and Replication Program as Head of the Antiviral Immunity and Resistance Section. His recent awards include an NCI Director's Award (2023), an NIH Intramural Targeted Anti-COVID-19 Program Award (2020), and a Collaborative Project Award from the National Interagency Confederation for Biomedical Research (2018). He is currently on the Editorial Board of Viruses, Frontiers in Virology, and Review Commons and was appointed as Guest Editor for the Journal of Molecular Biology Special Issue on "Antiviral Innate Immunity." Dr. Compton currently serves as an Adjunct Instructor in the Department of Biology at Hood College, as a member of the COVID-19 Scientific Interest Group Steering Committee, and as a CCR Tenure Track Investigator Representative.
Job Vacancies
We have no open positions in our group at this time, please check back later.
To see all available positions at CCR, take a look at our Careers page. You can also subscribe to receive CCR's latest job and training opportunities in your inbox.
Team





News
NIH Fellows Award for Research Excellence
Mahesh Agarwal, postdoc in the Compton Lab, was recognized with a 2024 NIH Fellows Award for Research Excellence (FARE). Congrats, Mahesh!
NIH Intramural AIDS Research Fellowship Award
Mahesh Agarwal, postdoc in the Compton Lab, was selected to receive the 2023 NIH Intramural AIDS Research Fellowship (IARF) Award for his proposal to characterize novel functions of the HIV-1 accessory protein, Nef. Congrats, Mahesh!
Discovery in Compton Lab featured in NIH Catalyst Magazine
Work published by first author Scarlett Shi in Nature Communications entitled "Omicron Spike confers SARS-CoV-2 with enhanced infectivity and interferon resistance in human nasal tissue" was highlighted in NIH Catalyst Magazine in March 2024. Read the coverage here.
DRP Think Tank Travel Award
Abbie Jolley, postbac in the Compton Lab, was selected to received a travel award in recognition of her excellent talk delivered at the DRP Think Tank meeting in 2023. Congrats, Abbie!
Discovery in Compton Lab featured in NIH Catalyst Magazine
Work published by first author Scarlett Shi in Journal of Clinical Investigation entitled "Rapalogs downmodulate intrinsic immunity and promote cell entry of SARS-CoV-2" was featured as a Research Brief in NIH Catalyst Magazine in January 2023. Read the coverage here.
Recent Editorial Appointments for Alex Compton
Alex Compton was recently appointed to the following editorial positions:
- Guest Editor, Viruses Special Issue on "Interferon-Induced Transmembrane Proteins," 2023
- Associate Editor, Frontiers in Virology, 2021
- Guest Editor, Journal of Molecular Biology Special Issue on “Antiviral Innate Immunity,” 2021
- Editorial Board Member, Viruses, 2020
- Editorial Advisory Board Member, Review Commons (European Molecular Biology Organization), 2020
NIH Fellows Awards for Research Excellence
NIH Fellows Award for Research Excellence (FARE) was granted to Kazi Rahman in 2022, Charles (Chad) Coomer in 2021 and 2019, and Scarlett (Guoli) Shi in 2018. These awards, which acknowledge outstanding scientific research performed by intramural postdoctoral fellows or graduate students, are based on scientific merit, originality, experimental design, and overall quality and presentation of the abstracts.
NIH Graduate Student Research Award
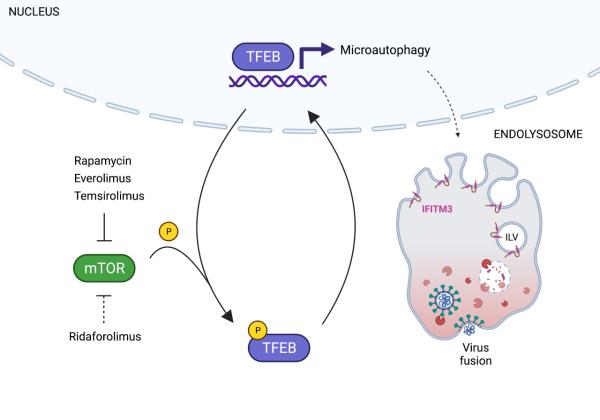
Charles (Chad) Coomer received an NIH Graduate Student Research Award for his presentation on “The Functional Relationship between Antiviral Restriction Factor IFITM3 and Cellular Cholesterol Homeostasis” at the 2020 NIH Graduate Student Research Symposium: Faces of Tomorrow’s Science.
NIH Intramural Targeted Anti-COVID-19 Program Award
Alex Compton received an NIH Intramural Targeted Anti-COVID-19 (ITAC) Program Award in 2020 to support his research project on “Deciphering the Double-Edged Role of IFITM3 During SARS-CoV-2 Infection.” ITAC awards are administered by the NIH Office of Intramural Research to provide intramural investigators with up to 2 years of funding for research into understanding and/or combatting COVID-19. Of the 159 applicants for this competitive funding, Dr. Compton was among the 40 investigators who received awards.
NIH Intramural AIDS Research Fellowship
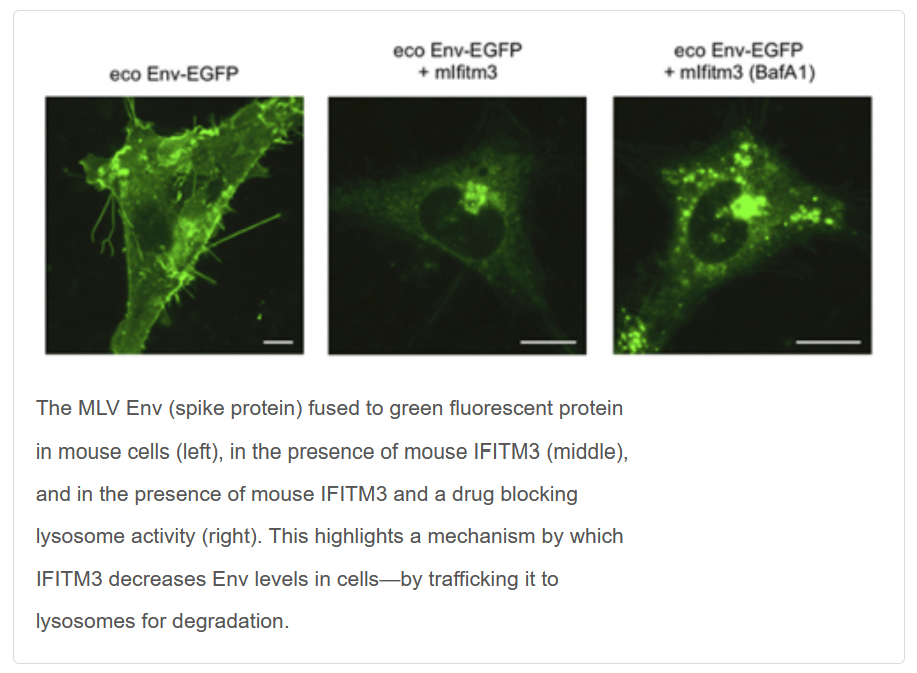
Charles (Chad) Coomer received an Intramural AIDS Research Fellowship (IARF) in 2020 to support his research project on "Revealing the Mechanism IFITM3-Mediated Inhibition of HIV-1 Envelope Using Advanced Fluorescence and Super-Resolution Microscopy." Awarded by the Office of AIDS Research, Office of Intramural Research, and Office of Intramural Research & Training in the National Institutes of Health, IARFs include full stipend support to successful candidates who demonstrate outstanding scientific potential through both an imaginative and thoughtful research plan and a well thought out career development plan.
Sallie Rosen Kaplan Postdoctoral Fellowship for Women Scientists
Saliha Majdoul (1st on left in photo below) was awarded a 2020 Sallie Rosen Kaplan (SRK) Postdoctoral Fellowship for Women Scientists at the National Cancer Institute. The SRK Fellowship is a highly competitive annual program that provides additional mentoring opportunities, networking, seminars, and workshops to help prepare NCI’s female postdoctoral fellows for the competitive nature of the job market and help them to transition to independent research careers. The highlight of this selective program is a 30-week course entitled "Career Building for Women in Science," which includes two day-long workshops. The SRK Fellowship also includes mentoring opportunities with successful women scientists from government, industry, and academia.

{kind=link}
Interview with Compton and Rein Labs in NCI-Frederick Poster
[The following excerpt is from the article "Two Teams, One Lab, One Partnership to Find Vulnerabilities in Viruses," published by Samuel Lopez in the NCI at Frederick Poster, 29 June 2020.]
Sharing a lab with another team may sound like an inconvenience to some, like the professional equivalent of sharing a bedroom with a sibling.
But not for Alex Compton, Ph.D.; Alan Rein, Ph.D.; and their colleagues. In their case, sharing a lab led to a rich collaboration, mentoring opportunities, and a study that uncovered facets of how cells interact with viruses.
The partnership began nearly three years ago, when Compton joined the HIV Dynamics and Replication Program at NCI at Frederick as head of the Antiviral Immunity and Resistance Section. His small team was given space in a laboratory partially occupied by Rein’s team, the Retrovirus Assembly Section. Soon, the scientists were sharing ideas and asking questions that required their combined expertise to answer.
“[My team and I] were working on aspects of IFITM function and basic questions. [Alan] simply asked whether they could inhibit [murine leukemia virus], as that happens to be his favorite virus model,” Compton said.
While simple, the question had far-reaching implications for scientists’ understanding of how viruses infect cells and spread in a host. IFITM (interferon-induced transmembrane) is a group of proteins that hinder viral infections in human and animal cells, a sort of cellular immune system. Murine leukemia virus (MLV) is a retrovirus, like HIV and human T-cell lymphotropic virus, that causes leukemia in mice by establishing long-term infections that dodge the mice’s natural defenses.
Collaborative Project Award, National Interagency Confederation for Biomedical Research
Alex Compton received a Collaborative Project Award from the National Interagency Confederation for Biomedical Research (NICBR) in 2018 to support his project on “Understanding the Mechanism of IFITM-Mediated Restriction of Virus Infection Through Its Interaction with ZMPSTE24.” A consortium of Federal research partners, the NICBR includes elements of the Departments of Health and Human Services, Defense, Agriculture, and Homeland Security.