
Eric O. Freed, Ph.D.
- Center for Cancer Research
- National Cancer Institute
- Building 535, Room 110
- Frederick, MD 21702-1201
- 301-846-6223 (office)
- 301-846-6483 (lab)
- efreed@mail.nih.gov
RESEARCH SUMMARY
Dr. Freed is recognized as a leader in the field of virus assembly who has made important strides in understanding the mechanisms of retroviral replication at the molecular level, with an emphasis on late stages of the HIV-1 replication cycle. As Director of the HIV DRP, he oversees a program of basic, translational, and clinical research aimed at developing a better understanding of HIV that can be used to generate more-effective treatment and cure strategies and elucidating mechanisms of antiviral innate immunity and cancer virus replication. Dr. Freed's research focuses on HIV-1 Gag trafficking, Env incorporation, virus assembly, budding, release, maturation, and drug resistance. Dr. Freed has a special interest in the complex relationship between viral proteins and cellular factors and pathways, believing that characterizing fundamental aspects of the retrovirus replication cycle will suggest novel targets for the development of antiretroviral therapies. Recent work in the Freed lab has also focused on the ability of Env mutations to broadly rescue defects in virus replication, including those conferred by antiretrovirals.
Areas of Expertise
Eric O. Freed, Ph.D.
Research
Assembly and Release of HIV-1 and Other Retroviruses
Retroviral Gag proteins are synthesized in the cytoplasm of the infected cell and assemble into virus particles that typically bud from the plasma membrane (PM). Expression of Gag proteins alone is generally sufficient for the assembly and release of noninfectious, virus-like particles (VLPs). The mature Gag proteins [matrix (MA), capsid (CA), and nucleocapsid (NC)] are generated concomitant with virus release upon cleavage of the Gag precursor by the viral protease (PR) [Freed, Nat. Rev. Microbiol. 2015]. PR-mediated Gag processing leads to virus maturation, a morphological transformation essential for virus infectivity. Retroviral gag genes often encode other domains and spacer peptides in addition to MA, CA, and NC. For example, the HIV-1 Gag precursor includes two spacer peptides (SP1 and SP2) and the p6 domain.
Retroviral Gag trafficking and assembly. After Gag synthesis, the MA domain directs Pr55Gag to the PM. The affinity of the MA domain for membrane is provided in part by a myristic acid moiety covalently attached to the N-terminus of MA. Sequences in MA downstream of the myristate also contribute to membrane binding—in particular, a highly basic patch of amino acid residues.
A large body of data indicates that HIV-1 assembly takes place predominantly at the PM [Freed, Nat. Rev. Microbiol. 2015]. Although the cellular determinants that regulate the site of HIV-1 assembly remain to be fully defined, we demonstrated that lipid rafts serve as sites for assembly at the PM [Ono & Freed, PNAS 2001] and identified the phosphoinositide lipid phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2] as a host factor involved in directing Gag to the PM [Ono et al., PNAS 2004]. We are also working to elucidate the role of inositol-hexakisphosphate (IP6) in HIV-1 assembly and maturation [Mallery et al., Cell Rep. 2019; Mallery et al., Sci. Adv. 2021; Renner et al., Nat Struct Mol Bio. 2023; Kleinpeter et al., J Mol Bio.2023] and have discovered that the cellular protein PSGL-1, when incorporated into virus particles, blocks the ability of those particles to bind to target cells [Fu et al., PNAS 2020]. We have recently discovered that the host cell enzyme neutral sphingomyelinase 2, which functions in the ceramide biosynthetic pathway, is required for the maturation of HIV-1 and other primate lentiviruses. Inhibitors of nSMase2 disrupt HIV-1 replication both in vivo and in humanized mouse models for HIV-1 infection [Waheed et al., PNAS 2023; Yoo et al., PNAS 2023].
Env incorporation and function. The envelope (Env) glycoproteins of HIV-1 are synthesized as a polyprotein precursor, gp160, that is proteolytically processed by the cellular protease furin to generate the surface (SU) glycoprotein gp120 and the transmembrane (TM) glycoprotein gp41 [Checkley et al., J. Mol. Biol. 2011]. Incorporation of Env glycoproteins into virions is an essential step in the replication process; however, the mechanism by which the Env glycoproteins are incorporated remains incompletely characterized. Several lines of evidence suggest that HIV-1 Env glycoproteins are actively recruited into virions via direct interactions between Env and MA; for example, mutations in both the MA domain of Gag and the cytoplasmic tail of gp41 can block HIV-1 Env incorporation. We showed that in most cell lines and in primary cell types, gp41 truncations severely disrupt Env incorporation but that in some cell lines these gp41 truncation mutants are efficiently incorporated [Murakami & Freed, PNAS 2000]. The cell-type-dependent requirement for the gp41 cytoplasmic tail in Env incorporation hints at the involvement of host cell factors. However, the identity of such factors remains to be defined. We are currently using a range of cell biology, virology, biochemical, and imaging approaches to characterize viral and cellular determinants of Env incorporation. Our findings highlight a requirement for trimerization of the MA domain of Gag in Env incorporation [Tedbury et al., PNAS 2016; Tedbury et al., J. Virol. 2020]. Our ongoing work has demonstrated that the cellular MARCH family of E3 ubiquitin ligases restricts the expression and incorporation of a range of viral glycoproteins including HIV-1 Env, VSV-G, Ebolavirus GP, and the spike protein of SARS-CoV-2 [Lun et al., mBio 2021].
More than two dozen drugs, most of which target the viral enzymes reverse transcriptase (RT), protease (PR), or integrase (IN), are currently available to treat HIV-1-infected individuals. These drugs, when used in combination (combination antiretroviral therapy or cART), effectively suppress virus replication in most treated individuals. However, resistance to cART does arise in some individuals, particularly in the context of poor adherence, suboptimal drug regimens, or lack of viral load monitoring. Resistance is usually associated with mutations in the viral genes targeted by antiretrovirals (ARVs). However, in some cases, resistance arises without mutations in the drug-target gene. For example, many patients fail PR inhibitor (PI)-based therapy without acquiring mutations in PR, and in a number of individuals treated with the highly potent IN strand-transfer inhibitors (INSTIs) dolutegravir (DTG) and raltegravir (RTG), patients fail therapy without the virus acquiring mutations in IN. We performed selections for DTG resistance and identified Env mutations that conferred resistance to DTG. Resistance was linked to the ability of the Env mutations to significantly enhance virus spread through cell-to-cell contact. These results demonstrate that, at least in vitro, mutations in Env can confer resistance to an ARV [Van Duyne et al., PNAS 2019]. Recent work has demonstrated that the Env mutations can confer resistance not only to the INSTI DTG but also to RT and PR inhibitors [Hikichi et al., mBio 2021]. Ultimately, we plan to fully define the mechanism by which the identified mutations in Env confer escape from ARVs and determine whether Env-mediated escape also occurs in vivo.
Retrovirus budding. The p6 domain of HIV-1 Gag is required for efficient virus budding. We mapped the virus release function of p6 to a highly conserved Pro-Thr-Ala-Pro (PTAP) motif near the N-terminus of p6 [Huang et al., J. Virol. 1995; Demirov et al., J. Virol. 2002; Fujii et al., Nat. Rev. Microbiol. 2007]. Other retroviral Gag proteins also possess "late domains" that, like the PTAP motif of p6, promote virus release.
Numerous lines of evidence support the hypothesis that retroviral late domains function by interacting with host factors—specifically, components of the cellular endosomal sorting complex required for transport (ESCRT) machinery [Freed, Nat. Rev. Microbiol. 2015]. The ESCRT machinery is composed of four multiprotein complexes (ESCRT-0, I, II, and III) and a variety of accessory proteins including Alix and the ATPase Vps4. This cellular apparatus functions in the sorting and release of cargo proteins into vesicles that bud into late endosomes and also plays a key role in the abscission step of cytokinesis. Retroviruses have evolved to usurp this cellular budding machinery to promote their release from the PM. Work from several labs, including ours [Demirov et al., PNAS. 2002], demonstrated that HIV-1 recruits the ESCRT machinery primarily through a direct interaction between the PTAP motif in p6 and the ESCRT-I component tumor susceptibility gene 101 (Tsg101). Secondary interactions occur between p6 and Alix.
Virion maturation. PR-mediated cleavage of the Gag and Gag–Pol precursors leads to a dramatic change in virion morphology, a process known as maturation. The highly ordered nature of Gag processing and the strict dependence on complete processing for proper virion maturation make the Gag processing cascade a potential target for drug development. Indeed, the betulinic acid derivative dimethylsuccinyl betulinic acid [PA-457 or bevirimat (BVM)] potently inhibits HIV-1 infectivity by targeting a late Gag processing event: the cleavage of the CA–SP1 processing intermediate to mature CA [Li et al., PNAS 2003]. By specifically disrupting this step in Gag processing, BVM treatment leads to the formation of poorly infectious viral particles with aberrantly condensed cores. We have selected and characterized a number of single amino acid mutations in the CA–SP1 boundary region that confer resistance to BVM, establishing this region of Gag as the target of the inhibitor [Adamson et al., J. Virol. 2006]. Clinical trials conducted with BVM provided mixed results; in some patients significant reductions in viral loads were achieved, whereas in other patients no benefit of BVM therapy was observed. We and others were able to attribute this lack of response to polymorphisms in SP1. We also performed studies on a second HIV-1 maturation inhibitor, PF-46396, developed by Pfizer. While structurally distinct from BVM, PF-46396 also inhibits CA–SP1 processing. Particularly interesting is our observation that a number of PF-46396-resistant mutants are severely defective for assembly and infectivity in the absence of the compound but produce infectious virions in its presence [Waki et al., PLoS Pathog. 2012]. With collaborators at DFH Pharma, we have identified a series of highly potent BVM derivatives that display broad activity against strains of HIV-1 that are resistant to BVM [Urano et al., Antimicrob. Agents Chemother. 2016; Urano et al., J. Virol. 2019]. We have also been working with structural biologists to solve the structure of maturation inhibitors bound to their target in Gag [Sarkar et al., NatComm 2023]. This work advances our efforts to develop virus maturation inhibitors as an antiviral strategy.
Publications
- Bibliography Link
- View Dr. Freed's PubMed Summary
- View Dr. Freed's Google Scholar page
Neutral sphingomyelinase 2 is required for HIV-1 maturation.
HIV-1 is dependent on its immature lattice to recruit IP6 for mature capsid assembly.
Mutations in the HIV-1 envelope glycoprotein can broadly rescue blocks at multiple steps in the virus replication cycle.
Biochemical evidence of a role for matrix trimerization in HIV-1 envelope glycoprotein incorporation
Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane
Biography

Eric O. Freed, Ph.D.
Dr. Eric Freed received his Ph.D. in 1990 in the laboratories of Drs. Rex Risser and Howard Temin at the University of Wisconsin-Madison and did postdoctoral work with Dr. Temin at UW-Madison in 1991. His work in Madison focused on the function of the murine leukemia virus and HIV envelope glycoproteins in membrane fusion and virus entry. He joined the Laboratory of Molecular Microbiology at the National Institute of Allergy and Infectious Diseases (LMM/NIAID) in 1992, where he worked with Dr. Malcolm Martin on HIV assembly and entry/post-entry events in the HIV replication cycle. In 1997 Dr. Freed was appointed as a Tenure-Track Investigator in LMM/NIAID, and he was promoted to a tenured Senior Investigator position in 2002. In 2003 he joined the HIV Drug Resistance Program (HIV DRP, renamed the HIV Dynamics and Replication Program in 2015) as Head of the Virus-Cell Interaction Section and was appointed to the NCI Senior Biomedical Research Service in 2011. Dr. Freed was appointed Deputy Director of the HIV DRP in 2014 and Director of the Program in 2015. He was an Organizer of the 2004 Cold Spring Harbor Retroviruses Meeting, 2006 ASCB Conference "Cell Biology of HIV-1 and Other Retroviruses," 2012 Keystone Symposium "Frontiers in HIV Pathogenesis, Therapy and Eradication," 2014 Keystone Symposium "The Ins and Outs of Viral Infection: Entry, Assembly, Exit and Spread," Viruses 2016 Conference "At the Forefront of Virus–Host Interactions," (Basel, Switzerland) Viruses 2018 Conference "Breakthroughs in Virus Replication," (Barcelona, Spain), 2018 gp41 Cytoplasmic Tail Structure and Function Workshop, Viruses 2020 Conference “Novel Concepts in Virology” (Barcelona), Viruses 2022 Conference “At the Leading Edge of Virology Research” (virtual), and Viruses 2024 Conference “A World of Viruses” (Barcelona). He also served on the Scientific Committee of the International Retroviral Nucleocapsid Protein and Assembly Symposium in 2013, 2016, 2019, 2023, and 2025 and the Organizing Committee of the Annual Meeting of the American Society for Virology in 2018. He has served as Co-Organizer of the Annual HIV DRP Conference and Annual David Derse Memorial Lecture and Award since 2012 and Organizer of the Annual George Khoury Memorial Lecture since 2014. Dr. Freed is the founding Editor-in-Chief of Viruses and was appointed Editor of Journal of Molecular Biology in 2012. He also currently serves on the Editorial Boards of a number of journals, including Journal of Virology, Retrovirology, and Frontiers in Virology, and is an Associate Editor for Science Advances and Fields Virology. He was a sitting member of the NIH AIDS Discovery and Development of Therapeutics (ADDT) study section (2012-2017) and served as ADDT Chair from 2015 to 2017. He is Co-Director of the University of Maryland Virology Program and an Adjunct Professor in the Department of Cell Biology and Molecular Genetics at the University of Maryland, College Park and Adjunct Professor at the University of Delaware. Dr. Freed is a member of the Pittsburgh Center for HIV Protein Interactions (PCHPI) and the Behavior of HIV in Viral Environments (B-HIVE) Center and is a member of the Scientific Advisory Board of the Center for Structural Biology of HIV RNA (CRNA). Dr. Freed received the NCI Mentor of Merit Award in 2010 and the NCI Women Scientists Advisors Mentoring and Leadership Award in 2021 for excellence in mentoring and guiding the careers of scientists in all stages of their careers. He received the Outstanding Science Alumni Award from Penn State University in 2014 and the NCI Research Highlights Award in 2016. In recognition of his outstanding contributions to the field of retrovirology, Dr. Freed was awarded the KT Jeang Retrovirology Prize in 2018 and the Ohio State University Center for Retrovirus Research Distinguished Research Career Award in 2020. He was elected to Fellowship in the American Academy of Microbiology in 2019. He delivered a plenary lecture at the 2024 Conference on Retroviruses and Opportunistic Infections (CROI) and a keynote address at the 2024 Cold Spring Harbor Retroviruses Meeting.
Job Vacancies
We have no open positions in our group at this time, please check back later.
To see all available positions at CCR, take a look at our Careers page. You can also subscribe to receive CCR's latest job and training opportunities in your inbox.
Team











News
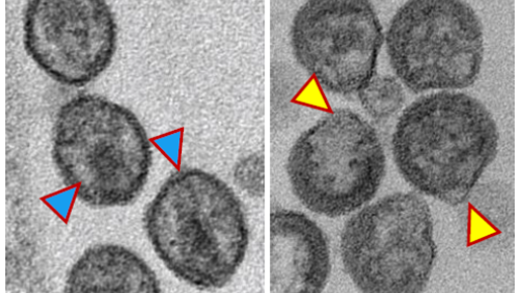
Alex Kleinpeter publishes in Nature Communications
Alex Kleinpeter publishes in the journal Nature Communications showing the structural basis for HIV-1 capsid adaption to a deficiency in IP6 packaging.
- Structural basis for HIV-1 capsid adaption to a deficiency in IP6 packaging. Nat Commun 16, 8152 (2025)
Eric Freed and Alex Kleinpeter publish a chapter in "Principles and Practices of Infectious Diseases"

NIH Fellows Awards for Research Excellence
Yuta Hikichi won 2026 NIH Fellows Awards for Research Excellence (FARE) for travel to attend and present his work at a scientific meeting in the U.S. This award, which acknowledges outstanding scientific research performed by intramural postdoctoral fellows, is sponsored by the NIH Fellows Committee, Scientific Directors, and Office of Intramural Training and Education and is funded by the Scientific Directors. FARE awards are based on scientific merit, originality, experimental design, and overall quality/presentation of the abstracts. Members of the Freed Lab who were FARE awardees in previous years include, Yuta Hikichi (2022), James Kirui (2022), Rachel Van Duyne (2015), Lillian Kuo (2012), Kayoko Waki (2012), Angelica Martins (2012), Muthukumar Balasubramaniam (2010), Benjamin Luttge (2010), Catherine Adamson (2009), and Karine Gousset (2007).
Freed lab at CSHL Retrovirology Meeting 2025
Members of the Freed lab attended the 2025 Cold Spring Harbor Laboratory Retrovirology Meeting.
Yuta Hikichi and Alex Kleinpeter were selected to give oral presentations. Abdul Waheed gave a poster presentation.

Intramural AIDS Research Fellowships
Intramural AIDS Research Fellowship (IARF) awards from the Office of AIDS Research, Office of Intramural Research, and Office of Intramural Research & Training in the National Institutes of Health include full stipend support to successful candidates who demonstrate outstanding scientific potential through both an imaginative and thoughtful research plan and a well thought out career development plan.
Rebecca Grande received an IARF award in 2025 to support her research project.
The following postdoctoral fellows in the Freed Lab received IARF awards in previous years: Geraldine Vilmen (2023), Hana Veler (2022, 2023), Yuta Hikichi (2021, 2022), Alex Kleinpeter (2020, 2021), Cheng man (Bonnie) Lun (2019, 2020), Melissa Fernandez (2018, 2019), Mariia Novikova: "Characterization of Antiretroviral Activity of Second-Generation Maturation Inhibitors and Mechanisms of Resistance" (2017), Rachel Van Duyne: "Challenging the Paradigm for Antiretroviral Resistance through the Study of Non-Canonical HIV-1 Escape Mutants" (2017), Emiko Urano: "Development of Potent and Broadly Active HIV-1 Maturation Inhibitors" (2016), Mariia Novikova: "Mechanisms of HIV-1 Gag Lattice Formation and Env Incorporation" (2016), Rachel Van Duyne: "Characterizing the Host Cell Factors Involved in HIV-1 Gag Trafficking to Sites of Virus Assembly" (2015), Robert Buckheit: "The Effect of Host Antiviral Restriction Factors on Viral Budding and Maturation" (2014), Lillian Kuo: "Characterizing the Role of HIV-1 p6-Alix Binding in HIV-1 Cell-to-Cell Infectivity" (2011, 2012), and Philip Tedbury: "HIV-1 Gag in Assembly and Release: Interactions with gp41 and Cellular Host Factors" (2011, 2012)
Poster Presentations at NIH Summer Poster Day 2025
Ellie Abbot, Diego De Nault, and Thomas Lidhal showcased their research to the NIH community at the summer poster day in Bethesda (2025). NIH Postbac Poster Day winners, Ellie Abbot and Diego De Nault were recognized as outstanding based on high scores they received during the judging process.

Yuta Hikichi publishes in Science Advances
Yuta Hikichi and Hana Veler publish in the journal Science Advances showing that structural maturation of the matrix lattice is not required for HIV-1 particle infectivity
Structural maturation of the matrix lattice is not required for HIV-1 particle infectivity. SciAdv. May 9;11(19):eadv4356.
Poster Presentations at Spring Festival Poster Blitz, Ft. Detrick 2025
Postbacs Ellie Abbot, Diego De Nault, and Thomas Lidhal presented at the Spring Festival Poster Blitz. Thomas Lidhal won an excellent poster award!

Rebecca Grande elected as chair of DRPAC
Rebecca Grande will take over from the outgoing chair Alex Kleinpeter. The DRPAC was established in 2023 and is made up of representatives from each of the DRP labs. DRPAC subcommittees help integrate new hires, facilitate external and internal communications and organize professional and social events, seminars and conferences.
Freed lab publishes in mBio
Alex Kleinpeter, Meghna Tripathi and Binh Nguyen contribute to a publication in the journal mBio showing the structural and mechanistic bases for resistance of the M66I capsid variant to lenacapavir .
- Structural and mechanistic bases for resistance of the M66I capsid variant to lenacapavir. mBio. 2025 May 14: e0361324
Freed lab publishes in mBio
Yuta Hikichi contributes to a publication in the journal mBio showing that a monomeric envelope glycoprotein cytoplasmic tail is sufficient for HIV-1 Gag lattice trapping and incorporation.
- A monomeric envelope glycoprotein cytoplasmic tail is sufficient for HIV-1 Gag lattice trapping and incorporation. mBio. 2025 April 15: e0210524
Travel Awards, HIV DRP Think Tank Meeting
Yuta Hikichi received a $1000 travel award from the HIV DRP for one of the two best presentations by NCI fellows at the 2025 HIV DRP Think Tank Meetings. Members of the Freed Lab who received travel awards in previous years include Alex Kleinpeter (2024), Rachel Van Duyne (2017, 2018), Mariia Novikova (2016), Emiko Urano (2015), Kayoko Waki (2011), and Muthukumar Balasubramaniam (2010)
Sallie Rosen Kaplan Postdoctoral Fellowship for Women Scientists
Rebecca Grande was awarded a 2025 Sallie Rosen Kaplan (SRK) Postdoctoral Fellowship for Women Scientists at the National Cancer Institute. The SRK Fellowship is a highly competitive annual program that provides additional mentoring opportunities, networking, seminars, and workshops to help prepare NCI’s female postdoctoral fellows for the competitive nature of the job market and help them to transition to independent research careers. The highlight of this selective program is a 30-week course entitled "Career Building for Women in Science," which includes two day-long workshops. The SRK Fellowship also includes mentoring opportunities with successful women scientists from government, industry, and academia.
Travel Awards, Fall HIV/AIDS & Cancer Virology Think Tank Meeting
Yuta Hikichi won a $1000 travel award for his outstanding oral presentation at the 2024 Fall HIV/AIDS & Cancer Virology Think Tank Meeting. In addition, Alex Kleinpeter won a $700 travel award for his outstanding posters at the 2024 Fall HIV/AIDS & Cancer Virology Think Tank Meeting. This annual Think Tank meeting on the NIH-Bethesda campus provides a venue for students, postdoctoral fellows, and staff scientists to present emerging work and hypotheses in the field of cancer virology. The Think Tank travel awards are provided by the Center of Excellence in HIV/AIDS & Cancer Virology, Center for Cancer Research, NCI. Members of the Freed Lab who received travel awards in previous years include Yuta Hikichi (2022, 2023), Lindsey Farr (2023), Binh Nguyen (2023), Alex Kleinpeter (2022), Mariia Novikova (2017, 2016) and Rachel Van Duyne (2016), Philip Tedbury (2014), Angelica Martins (2013), and Emiko Urano (2013).
Keynote Speaker Eric Freed at World AIDS Day 2024
Eric Freed - keynote speaker at World AIDS Day 2024, "Novel therapies to prevent and treat HIV infection". Available to watch here.
Hana Veler presents at the Virology Program Retreat 2024
Hana Veler delivered a talk on her recent mBio paper "Guanylate binding protein 5 antagonizes viral glycoproteins independently of furin processing" at the University of Maryland - College Park Virology Program Retreat (2024).
Eric Freed delivers a talk at Malcolm A. Martin's Retirement Symposium 2024
Eric Freed delivered a talk "Mutations Outside Integrase Confer Resistance to Integrase Strand Transfer Inhibitors". Available to watch here.
Alex Kleinpeter publishes in Nature Communications
Alex Kleinpeter publishes in the journal Nature Communications, showing HIV-1 adapts to lost IP6 coordination through second-site mutations that restore conical capsid assembly (2024).
- HIV-1 adapts to lost IP6 coordination through second-site mutations that restore conical capsid assembly. Nat Commun. 2024 Sep 13;15(1):8017.
Eric Freed publishes in Viruses
In collaboration with colleagues at the American University, ViiV Healthcare, and District of Columbia Center for AIDS Research, Eric Freed publishes a review in the journal Viruses titled "Exploring HIV-1 Maturation: A New Frontier in Antiviral Development" (2024).
Exploring HIV-1 Maturation: A New Frontier in Antiviral Development. Sep 6;16(9):1423. PMID: 39339899.
Hana Veler publishes in mBio
Hana Veler, Abdul Waheed and Cheng Man Lun publish in the journal mBio, showing Guanylate binding protein 5 antagonizes viral glycoproteins independently of furin processing (2024).
- Guanylate-binding protein 5 antagonizes viral glycoproteins independently of furin processing. mBio.
Congratulations Geraldine Vilmen!
Geraldine Vilmen (visiting fellow) took up the position of Assistant Professor within The School of Natural Science and Mathematics at Mount St. Mary’s University, MD. Her research at Mount St. Mary’s will focus on HIV and the application of lentiviral research to cancer therapy. We wish you all the best!

Poster Presentations at NIH Summer Poster Day 2024
Nipun Gorantla (summer intern) mentored by Yuta Hikichi showcased his research to the NIH community at the summer poster day in Bethesda (2024).
Poster Presentations at NCI Frederick Summer Poster Day 2024
Aria Haghighat (summer intern) mentored by Alex Kleinpeter showcased his research to the wider NCI Frederick community at the summer poster day (2024).
Poster Presentations at the NIH HIV Structural Biology Meeting 2024
Yuta Hikichi, Alex Kleinpeter and Abdul Waheed presented posters at the 2024 HIV Structural Biology Meeting at NIH-Bethesda. A VideoCast is available here.
Federal Technology Transfer Award 2024
Binh Nguyen, Alex Kleinpeter and Eric Freed receive a 2024 Federal Technology Transfer Award (FTTA).
The awards are for those employees who contribute their skills, energy, and expertise to the advancement of the public health and science through technology transfer.
Freed lab at CSHL Retrovirology Meeting 2024
Members of the Freed lab attended the 2024 Cold Spring Harbor Laboratory Retrovirology Meeting.
Eric Freed gave the keynote address "Uncovering the mysteries of HIV replication (or trying to)". Available to watch here.
Alex Kleinpeter delivered his talk as the recipient of The Eighteenth Andy Kaplan Award on "Elucidating the role of IP6 in HIV-1 assembly, maturation, and post-entry events".
Yuta Hikichi was selected to give an oral presentation. Poster presentations were made by Sherimay Ablan, Lindsay Farr, Hana Veler, and Geraldine Vilmen.

Photo of Freed lab (with others) at CSHL.
Alex Kleinpeter wins The Andy Kaplan Prize, 2024
Congratulations to Alex Kleinpeter for winning the eighteenth Andy Kaplan Prize for his research work on “Elucidating the role of IP6 in HIV-1 assembly, maturation, and post-entry events” (2024).
The Andy Kaplan Prize was established in 2007, to be awarded annually, with the purpose of advancing the career and honoring the accomplishments of a distinguished postdoctoral scientist as he or she embarks on an independent career in retrovirology.
Eric Freed delivers plenary talk at the Conference on Retroviruses and Opportunistic Infections
Eric Freed delivered his plenary talk on “HIV Assembly, Maturation Inhibitors, and Drug Resistance” (2024).
New Investigator Scholarships, Conference on Retroviruses and Opportunistic Infections
Hana Veler presented a poster at the 2023 Conference on Retroviruses and Opportunistic Infections (CROI). Binh Nguyen was awarded a New Investigator Scholarships to present his research findings at CROI.
CROI scholarship awardees in previous years include Rachel Van Duyne (2018, 2017, 2019), Phuong Pham (2018, 2019), Mariia Novikova (2017, 2016, 2019), Justin Kaplan (2017), Emiko Urano (2016, 2014), Lillian Kuo (2013), and Catherine Adamson (2007).
Yuta Hikichi publishes in Science Advances
In collaboration with Yale University, Yuta Hikichi publishes in the journal Science Advances showing epistatic pathways can drive HIV-1 escape from integrase strand transfer inhibitors (2024).
- Epistatic pathways can drive HIV-1 escape from integrase strand transfer inhibitors. Sci Adv. 2024 Mar;10(9):eadn0042.
Abdul Waheed and colleagues at JHU publish two studies on nSMase2
In collaboration with John Hopkins University, Abdul Waheed, Eva Agostino, Lwar Naing and Yuta Hikichi publish papers in the journal PNAS showing a critical role of nSMase2, in the late stages of HIV-1 replication and maturation (2023).
- Neutral sphingomyelinase 2 is required for HIV-1 maturation. Proc Natl Acad Sci U S A. 2023 Jul 11;120(28):e2219475120.
- Inhibition of neutral sphingomyelinase 2 impairs HIV-1 envelope formation and substantially delays or eliminates viral rebound. Proc Natl Acad Sci U S A. 2023 Jul 11;120(28):e2219543120.
Alex Kleinpeter publishes in J Mol Biol
In collaboration with the University of Oxford, Imperial College London and The MRC Laboratory of Molecular Biology, Alex Kleinpeter and Sherimay D. Ablan publish findings in the Journal of Molecular Biology, that reinforce the importance of the six-helix bundle (6HB) in virus assembly, maturation, and infection and highlight the ability of IP6 to modulate 6HB stability (2023).
- The Effect of Inositol Hexakisphosphate on HIV-1 Particle Production and Infectivity can be Modulated by Mutations that Affect the Stability of the Immature Gag Lattice. J Mol Biol. 2023 Jun1;435(11):168037.
Pathway to Independence Award (K99/R00)
Alex Kleinpeter successfully competed for a K99/R00 Pathway to Independence (PI) Award from the National Institutes of Health in 2023. The PI Award Program establishes and maintains a strong cohort of new and talented, NIH-supported, independent investigators. This program is designed to facilitate a timely transition of outstanding postdoctoral researchers or clinician-scientists from mentored research positions to independent, tenure-track or equivalent faculty positions, and to provide independent NIH research support during the transition that will help these individuals launch competitive, independent research careers. Previous K99/R00 Award winner: Melissa Fernandez (2019)
Confirmed Talent Award from French Government
Geraldine Vilmen received an award from the French Government in December 2021 as a "confirmed talent" representing France overseas. She is part of an esteemed group of young people highlighting opportunities worldwide.
D.Phil. in Systems Approaches to Biomedical Science Awarded by University of Oxford
Hana Valer was awarded a D.Phil. degree in Biological and Biomedical Sciences by the University of Oxford in November 2021 after successfully defending her dissertation on the interaction between influenza A virus RNA-dependent RNA polymerase and cellular GTPase Rab11a. Her project was a collaboration between the Dunn School of Pathology and Division of Structural Biology (STRUBI) at the University of Oxford and the Diamond Light Source, the UK's national synchrotron science facility.
Eric Freed Received 2020 NCI Women Scientists Advisors Mentoring and Leadership Award
In recognition of his commitment to mentoring and his role in promoting diversity, Eric Freed received the 2020 NCI Women Scientists Advisors Mentoring and Leadership Award, which honors "exceptional dedication, leadership, and tireless efforts to promote and nurture NCI women scientists at all stages of their careers."
Eric Freed Received 2020 Distinguished Research Career Award from The Ohio State University Center for Retrovirus Research

Eric Freed was selected by the Center for Retrovirus Research of The Ohio State University to receive the 2020 Distinguished Research Career Award. This annual award honors the distinguished research career of a scientist working in the field of retrovirology. The retrovirologist is nominated by student and faculty members of the Center for Retrovirus Research and as part of the award recognition is invited to give a special lecture to all members of the Ohio State University biomedical research community. To read more about this award, click here.
NCI Director's Innovation Award
James Kirui received a 2020 NCI Director’s Innovation Award for his research proposal "Using highly sensitive reporter virus vectors to study late stages of HIV-1." With support from this award, Dr. Kirui developed innovative bioluminescent reporter virus vectors that provide a simple, rapid tool for functional studies of HIV-1 replication as well as high-throughput screening for cellular factors and small molecules that promote or inhibit HIV-1 particle production.
Travel Award, 39th Annual Meeting of the American Society for Virology (ASV 2020)
Melissa Fernandez was awarded an American Society for Virology Postdoctoral Scholar Travel Award for her abstract submission to ASV 2020 (meeting canceled due to COVID-19 pandemic).
Dr. Eddie Méndez Award
Melissa Fernandez was selected to be a recipient of the 2nd Annual Dr. Eddie Méndez Award, which recognizes outstanding postdoctoral scientists who are conducting research in cancer biology or infectious diseases. As an awardee, Dr. Fernandez presented her latest research findings at a scientific symposium in 2020 honoring Dr. Eddie Méndez and had the opportunity to discuss her work with faculty members of the Fred Hutchinson Cancer Research Center.
Postdoctoral Fellowship, Japan Society for the Promotion of Science
The Japan Society for the Promotion of Science (JSPS) awarded a 2020-2021 Research Fellowship to Yuta Hikichi for his project "Mutations in the HIV-1 envelope glycoprotein contribute to HIV drug resistance." The fellowship program sponsored by this society supports meritorious biomedical research projects undertaken in NIH laboratories by Japanese postdoctoral researchers. Emiko Urano was awarded a JSPS Research Fellowship from 2013 to 2015.
Lab Life
Goodbye Ellie!
The Freed lab said a warm farewell to Elizabeth "Ellie" Abbott (postbac) in August as she heads to grad school at Harvard University. Good luck Ellie!

We'll Miss You, Alex!
The Freed lab said a fond farewell to Alex Kleinpeter (postdoc) in May as he heads to the University of Iowa as a tenure-track assistant professor in the Department of Microbiology and Immunology at the University. We wish you all the best!

Farewell Lindsay!
The Freed lab said a fond farewell to Lindsay Farr (postbac) in July as she departs for grad school at UCSF. We wish you all the best!

Good luck Binh!
The Freed lab said a fond farewell to Binh Nguyen (postbac) in July as he departs for his MD/PhD program at Emory. We wish you all the best!

The Freed lab celebrating the holidays 2023

Members of the Freed lab enjoying a party hosted by Eric 2023

Members of the Freed lab enjoying the sights of Pittsburg whilst attending the Pittsburg Center for HIV Protein Interactions (PCHPI) 2023 Symposium

Left to right: Eric Freed, Binh Nguyen, Abdul Waheed, Lindsay Farr, Yuta Hikichi, Muhammad Behroz Khan
Freed Lab - March 2023

Left to right: Alex Kleinpeter, Eric Freed, Sherimay Ablan, Yuta Hikichi, Muhammad Behroz Khan, Hana Veler, Geraldine Vilmen, Binh Nguyen, Abdul Waheed, Lindsay Farr, Nidhi Chaudhary
Freed Lab — August 2019

Left to right: Melissa Fernandez, Sherimay Ablan, James Kirui, Cheng man (Bonnie) Lun, Alex Kleinpeter, Jennifer Simmons, Eric Freed, Lwar Naing, Ahlam Majadly, Abdul Waheed, Nicole Powell, Yuta Hikichi
Freed Lab — May 2018

Left to right: Hannah Carter, Sherimay Ablan, Rachel Van Duyne, Melissa Fernandez, Mariia Novikova, Cheng man (Bonnie) Lun, Ali Khan, Abdul Waheed, James Kirui, Phuong Pham, Eric Freed
Current Lab Members

Sherimay Ablan, B.A., Research Biologist
Sherimay earned her B.A. in biology from the University of Hawaii at Manoa. She contributes to all the research projects in the lab, with a primary focus on developing potent and broadly active HIV-1 maturation inhibitors (MIs) and elucidating the molecular, biochemical, and structural basis for MI activity. Sherimay received the Federal Technology Transfer Award for her work on MIs and the NIH Special Act or Service Award for her role in the design, planning, and renovation of a BSL2* HIV containment laboratory.

Yuta Hikichi, Ph.D., Postdoctoral Fellow (Visiting)
Yuta earned his Ph.D. in medical sciences from The University of Tokyo and AIDS Research Center, National Institute of Infectious Diseases, Japan, under the mentorship of Dr. Tetsuro Matano. His work is primarily focused on understanding the mechanism by which mutations in the HIV-1 envelope glycoprotein confer broad resistance to antiretrovirals. Yuta was awarded a Japan Society for the Promotion of Science Research Fellowship in 2020 and 2021, NIH Intramural AIDS Research Fellowship in 2021, and NIH Fellows Award for Research Excellence (FARE) in 2022.

Hana Veler, Ph.D., Postdoctoral Fellow (Visiting)
Hana was awarded a D.Phil. degree in biomedical sciences – pathology by the University of Oxford in November 2021 after successfully defending her dissertation on the interaction between influenza A virus RNA-dependent RNA polymerase and cellular GTPase Rab11a under Prof. Ervin Fodor’s mentorship. Hana’s project was a collaboration between the Dunn School of Pathology and Division of Structural Biology (STRUBI) at the University of Oxford and the Diamond Light Source, the UK's national synchrotron science facility. In the Freed lab, she is currently investigating how HIV-1 Gag cleavage/structural rearrangement of the matrix lattice during virion maturation affects envelope glycoprotein activation.

Abdul A. Waheed, Ph.D., Associate Scientist
Waheed received his Ph.D. in membrane biochemistry from the University of the Ryukyus, Okinawa, Japan. His current research is focused on host factors involved in HIV-1 replication and small-molecule inhibitors of HIV-1 replication. He was promoted to Staff Scientist in 2016 and Associate Scientist in 2019. In addition to mentoring all the student interns in the Freed lab, he has served in leadership roles in the CCR Staff Scientist/Staff Clinician Organization. Read more about Waheed in his CCR investigator profile.