Javed Khan, M.D.
- Center for Cancer Research
- National Cancer Institute
- Building 37, Room 2016B
- Bethesda, MD 20892
- 240-760-6135
- khanjav@mail.nih.gov
RESEARCH SUMMARY
The mission of the Oncogenomics Section is to harness the power of high-throughput genomics, proteomics, machine learning, and bioinformatics to improve the outcomes of children with high-risk metastatic, refractory, and recurrent cancers. The research goals are to integrate the data, decipher the biology of these cancers, identify and validate biomarkers and novel therapeutic targets, and rapidly translate our findings to the clinic.
Areas of Expertise
Javed Khan, M.D.
Research
Mission of the Oncogenomics Section
The mission of the Oncogenomics Section is to harness the power of high throughput genomic and proteomic methods to improve the outcome of children with high-risk metastatic, refractory and recurrent cancers. The research goals are to integrate the data, decipher the biology of these cancers and to identify and validate biomarkers and novel therapeutic targets and to rapidly translate our findings to the clinic.
1. Comprehensive Omics Analysis: a) Applying high-throughput genomics, proteomics mathematical modeling and bioinformatics to characterize currently incurable malignancies including metastatic, resistant and relapsed tumors for the identification and validation of biomarkers and therapeutic targets. b) Genome wide association studies and mutational screening of germ-line DNA.
2. Targeted Therapeutics: a) High-throughput siRNA, small molecule, natural products, and drug screening for high-risk pediatric malignancies. b) Development of molecularly targeted therapeutic agents against existing and newly identified targets.
3. Translational Oncology: Leverage the existing clinical and scientific strengths within the Genetics Branch including phase 1/2 therapeutics, immune/vaccine therapy and molecular biology to translate these findings to the clinic in an environment where there is state-of-the-art clinical care.
Publications
- Bibliography Link
- View Dr. Khan's Complete Bibliography at ResearcherID.
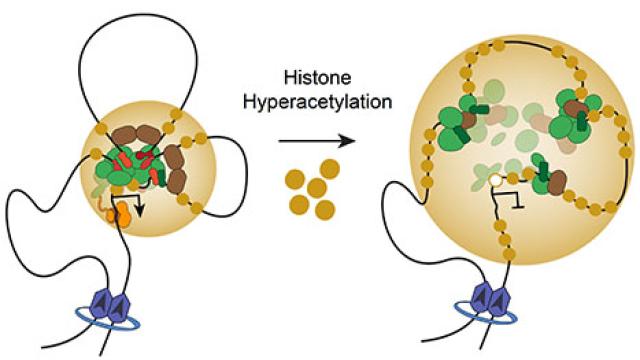
Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma
Chemical genomics reveals histone deacetylases are required for core regulatory transcription
PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability
MultiDimensional ClinOmics for Precision Therapy of Children and Adolescent Young Adults with Relapsed and Refractory Cancer: A Report from the Center for Cancer Research
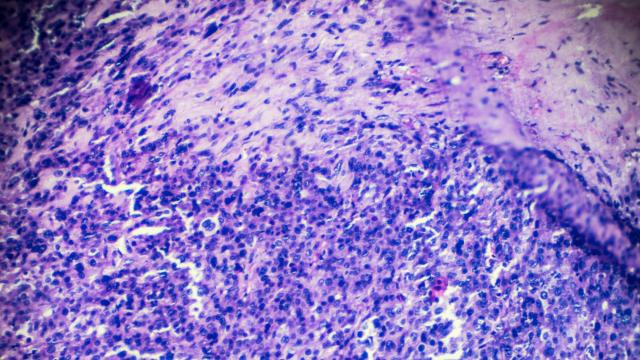
Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors
Biography

Javed Khan, M.D.
Dr. Khan obtained his bachelor's degree in 1984 and his master's degrees in 1989 in immunology and parasitology at England's University of Cambridge. He subsequently obtained his M.D. there and the postgraduate degree of MRCP (Membership of the Royal College of Physicians), equivalent to board certification in the United States. After clinical training in internal medicine and pediatrics as well as other specialties, he received a Leukemia Research Fellowship. In May 2001, Dr. Khan joined the Pediatric Branch, NCI, as a tenure track investigator. Dr. Khan and colleagues have published a new model for diagnosis of cancer using artificial neural networks (ANN), a form of artificial intelligence, and microarray technology. In April 2001, Dr. Khan was recognized by the American Association for Cancer Research for his work in tumor profiling by receiving a Scholar in Training Award. Recently Dr Khan has led an international collaboration to perform comprehensive analysis of pediatric cancer genomes using next generation sequencing strategies.
Job Vacancies
We have no open positions in our group at this time, please check back later.
To see all available positions at CCR, take a look at our Careers page. You can also subscribe to receive CCR's latest job and training opportunities in your inbox.