Cancer cells find ways to survive damage they cannot fix.
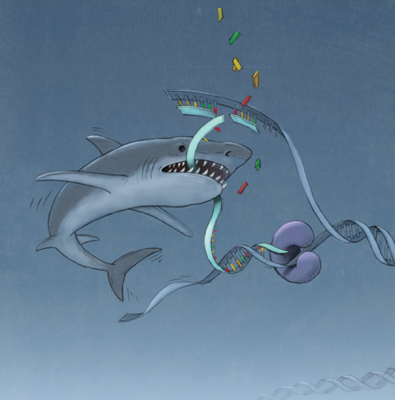
Nussenzweig and Sharan demonstrated how the replication fork is protected from predators. The image depicts MRE11 nuclease as a shark chewing up newly synthesized DNA at a replication fork, thus resulting in genome instability. Credit: Alan Hoofring, NIH
Damage to DNA is unavoidable. Chemicals, sunlight and routine cellular activities distort the genetic code by modifying its components and severing DNA strands. Most of the time, such damage has no harmful consequences because cells’ sophisticated DNA repair systems rush to replace the errors and mend the breaks. In some individuals, however, these repair systems are impaired by inherited genetic mutations and unrepaired damage accumulates, which increases the risk of tumors.
Mutations in the breast cancer susceptibility genes BRCA1 and BRCA2 predispose individuals to cancer. They also impair cells’ ability to sense and respond to DNA damage, a limitation that plays an important role in cancer therapy.
Patients whose cancers carry mutations in BRCA1 or BRCA2 almost always develop resistance to DNA-damaging chemotherapies. It has widely been assumed that this resistance develops as cells lacking functional BRCA1 or BRCA2 evolve new ways to repair their damaged DNA, but new work from CCR scientists shows that cancer cells lacking these repair factors can find other ways to thrive.
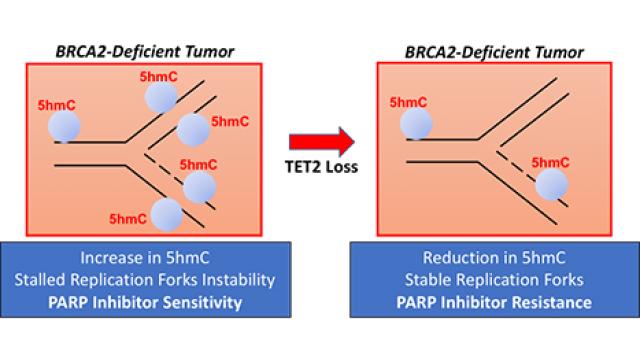
In a 2016 study reported in Nature, scientists led by André Nussenzweig, Ph.D., Chief of CCR’s Laboratory of Genome Integrity, discovered that cells with BRCA deficiencies can also become chemo-resistant by acquiring additional mutations that protect damaged DNA strands.
DNA double-strand breaks, which are caused by many chemotherapeutic agents and are the primary type of damage targeted by BRCA1 and BRCA2, normally stall the enzymes that copy DNA in preparation for cell division. This delay gives cells time to repair the damage. But in cells without BRCA2, the lesions go unrepaired, and DNA at the stall sites is degraded. Too many unrepaired lesions can ultimately lead to cell death. Nussenzweig and his colleagues found that when BRCA-deficient cells contain additional mutations that interfere with the DNA-degrading enzyme MRE11’s ability to find stall sites, they are able to survive double-strand breaks and become resistant to chemotherapeutic agents, such as cisplatin, that cause them.
In complementary work, a team led by Shyam Sharan, Ph.D., Deputy Director of CCR’s Mouse Cancer Genetics Program, found another challenge in treating BRCA-deficient cancer cells. Sharan and his colleagues investigated the effects of the cancer drug olaparib, which induces DNA damage and is toxic to BRCA-deficient cancer cells. However, when Sharan’s team tested the drug on cells with BRCA mutations that had not become cancerous, they found the opposite effect: Cells were able to keep growing despite their reduced ability to repair damaged DNA.
The survival mechanism Sharan’s team uncovered is similar to the drug-resistance strategy discovered by Nussenzweig’s group. They found that olaparib prevents the DNA-degrading enzyme MRE11 from accessing DNA sites where replication has stalled due to damage. This promotes cell survival and permits damaged DNA to be passed on to future generations of cells, which increases the risk of new tumors.
Sharan’s investigations of these basic cellular mechanisms, reported in Nature Communications, have important implications for the clinical use of olaparib and related drugs because they suggest that when these inhibitors are used to eliminate cancer cells in patients with inherited BRCA2 mutations, they may also increase the risk that new tumors will develop.
Chaudhuri AR, et al. Nature. 2016 Jul 20;535(7612):382-7.
Ding X, et al. Nat Commun. 2016 Aug 8;7:12425.