
Shiv Grewal, Ph.D.
- Center for Cancer Research
- National Cancer Institute
- Building 37, Room 6066
- Bethesda, MD 20892
- 240-760-7553
- grewals@mail.nih.gov
RESEARCH SUMMARY
Shiv Grewal's pioneering research has significantly advanced our understanding of genome organization and stability, with profound implications for cancer biology. Over more than two decades, his work has elucidated mechanisms of heterochromatin assembly and epigenetic control, offering fundamental insights into how disruptions in these processes contribute to genomic instability and cancer development. Grewal has demonstrated that distinct histone methylation patterns differentiate heterochromatin (closed) and euchromatin (open) regions of the genome. He was the first to establish that heterochromatic gene silencing can be stably propagated through meiosis and inherited in cis, extending the concept of inheritance beyond DNA sequences and defining fundamental principles governing epigenetic inheritance of silenced chromatin domains. A major breakthrough in his research identified a highly conserved link between RNA interference (RNAi) and heterochromatin assembly, reshaping our understanding of complex genome organization. This work was honored as "Breakthrough of the Year 2002" by Science magazine. The impact of his contributions is underscored by having three of his papers recognized by Nature as historic discoveries in the field of gene expression over the past 50 years. Currently, his lab focuses on uncovering how heterochromatin performs diverse regulatory functions to enhance genome stability, adaptive gene control, and three-dimensional genome organization.
Areas of Expertise
Shiv Grewal, Ph.D.
Research
Chromatin biology and epigenetics represent fundamental areas of molecular biology that have far-reaching implications for human health and disease. Aberrant chromatin regulation is now recognized as a hallmark of various diseases, particularly cancer. Indeed, when the mechanisms controlling the formation and maintenance of heterochromatin, a repressive form of chromatin, break down, it can lead to inappropriate gene expression, genomic instability, and cellular transformation. Importantly, understanding these processes has opened new avenues for therapeutic intervention, as many cancer treatments now target chromatin-modifying enzymes and other epigenetic regulators.
My research focuses on understanding how the genome is regulated within cells, particularly through the formation and maintenance of heterochromatin. We have uncovered epigenetic mechanisms by which cells can silence specific genes and maintain this silencing across cell divisions, a process critical for normal development and cellular function. Epigenetic mechanisms allow specific gene expression outputs and involve DNA and histone modifications, including those associated with heterochromatin. Heterochromatin maintains global patterns of gene expression and protects genomic integrity by prohibiting recombination between repetitive DNA elements and promoting proper segregation of chromosomes. Defects in heterochromatin mechanisms can lead to chromosome abnormalities frequently linked cancer. Alterations in histone methylation patterns and aberrant heterochromatin formation can lead to inappropriate gene silencing or activation, contributing to tumor development and progression. Understanding these mechanisms provides crucial insights for developing targeted therapeutic approaches in cancer treatment.
For more than 20 years, my lab has been delineating highly conserved heterochromatin assembly pathways. We were the first to define the epigenetic profile of an entire eukaryotic genome. Our comprehensive analyses have revealed that methylation of histone H3 lysine 9 (H3K9me) is strictly localized across heterochromatin domains, whereas H3K4me is specific to euchromatic regions. We have described the establishment of heterochromatin domains through nucleation and spreading activities catalyzed by the read/write activity of the histone methyltransferase Clr4/Suv39h, and we uncovered that heterochromatin provides a versatile recruiting platform for orchestrating diverse genome functions. Importantly, we have discovered that heterochromatin can be inherited epigenetically in a self-templating manner and described the fundamental mechanisms that promote epigenetic inheritance. Our work also led to the discovery of RNA-based mechanisms of heterochromatin assembly, including RNAi that utilizes small interfering RNAs (siRNAs) to target heterochromatin. RNA-based mechanisms of heterochromatin assembly are conserved in higher eukaryotes and have broad implications for human biology and disease.
Excitingly, our current research has revealed that multiple pathways converge to enforce a single key feature crucial for epigenetic inheritance of heterochromatin, namely the critical density of tri-methylated histone H3 lysine 9 (H3K9me3) that is required for self-propagation. Our work suggests high levels of H3K9me3 are necessary to support an increased local concentration of Clr4/SUV39H to promote the read-write activity, facilitating self-templated inheritance of silenced domains. However, despite significant progress, much still remains to be discovered. Below are our current research areas.
Current Research Areas
Epigenetic Inheritance: How are silenced chromatin domains heritably maintained?: The assembly and maintenance of repressive heterochromatin domains is achieved through modification of histones, particularly H3K9me3 that is recognized and deposited by Clr4/SUV39h via its ability to both “read” and “write”. Using a carefully designed genetic system, we have identified several factors that ensure faithful inheritance of heterochromatin by suppressing histone turnover. We have also provided direct evidence that histones are carriers of epigenetic information and discovered that a critical density of H3K9me3 is required to provide an epigenetic template for the spreading and epigenetic inheritance of heterochromatin. This work elucidated a fundamental theme in which the activities of multiple silencing factors converge to maintain H3K9me3 density, which in turn promotes epigenetic inheritance of heterochromatin through both mitosis and meiosis. Our recent work has revealed that histone deacetylation, a conserved feature of heterochromatin domains, blocks SWI/SNF subfamily remodelers involved in chromatin unraveling, thereby stabilizing H3K9me3 modified nucleosomes, to promote heterochromatin propagation. Moreover, we have reported that heterochromatin domains are specialized replication zones, enriched in replisome components, that collaborate with a histone chaperone complex to transfer parental modified histones to daughter DNA strands to preserve epigenetic memory. By elucidating these mechanisms, we provided a molecular basis for understanding how epigenetic information is maintained and transmitted. This insight not only enhances our fundamental knowledge of chromatin biology but also has potential implications for understanding biological processes including gene regulation, cellular differentiation, and disease states associated with epigenetic dysregulation.
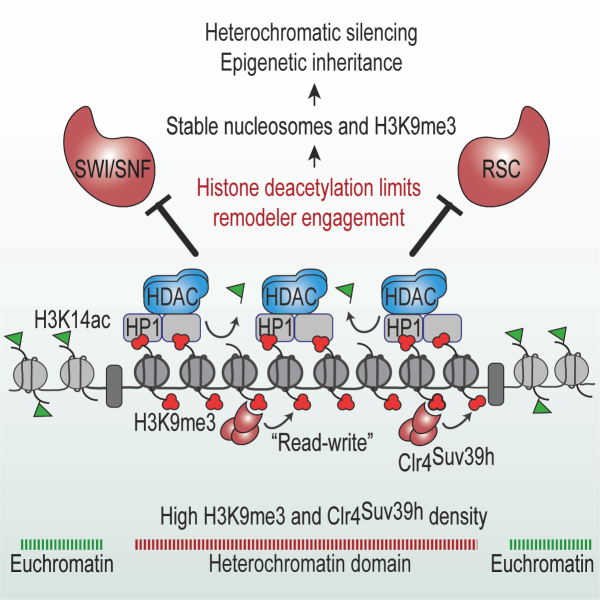
RNA-mediated Heterochromatin Assembly: My lab has discovered two distinct RNA-based mechanisms that direct the assembly of heterochromatin. We have described the role of the RNAi machinery and small RNAs in targeting heterochromatin (named “Breakthrough of the Year 2002” by Science magazine). We have also uncovered an RNAi-independent mechanism in which a nuclear RNA processing complex (named MTREC) cooperates with RNA polymerase II termination factors to direct the assembly of facultative heterochromatin domains under specific growth conditions. We are continuing to investigate mechanisms of heterochromatin assembly that are expected to have major implications for understanding gene regulatory processes in higher eukaryotes with implications for understanding how cancer cells can reactivate silenced tumor suppressor genes.
Signaling to Chromatin and Adaptive Genome Control: The complexity of eukaryotic genome regulation is evidenced by the ability to reprogram gene expression to adapt to environmental, nutritional or developmental signals. To this end, we are investigating how facultative heterochromatin domains can be modulated by signaling pathways. Our work has led to the finding that heterochromatin assembly factors are part of a rheostat-like buffering mechanism that limits the uncontrolled expression of a wide variety of genes in response to environmental change. TOR signaling pathway, a key nutrient sensor and regulator of cell growth in all eukaryotes, controls gene expression by targeting MTREC nuclear RNA elimination complex that promotes facultative heterochromatin and RNA decay to regulate gene expression. Two key findings are: (1) the RNA degradation/facultative heterochromatin assembly machinery is a central regulator of TOR-mediated control of cell proliferation and (2) TOR dynamically controls RNA elimination machinery to modulate facultative heterochromatin and coordinate developmental gene expression during gametogenesis, an extremely important process that ensures the genomic integrity of future generations and lies at the heart of many heritable human disorders. Moreover, the connection between TOR signaling and RNA elimination machinery has important implications for cancer biology, as dysregulation of TOR signaling is a hallmark of many cancers. We are continuing to explore how heterochromatin machinery intersects with signaling pathways to control gene expression programs.
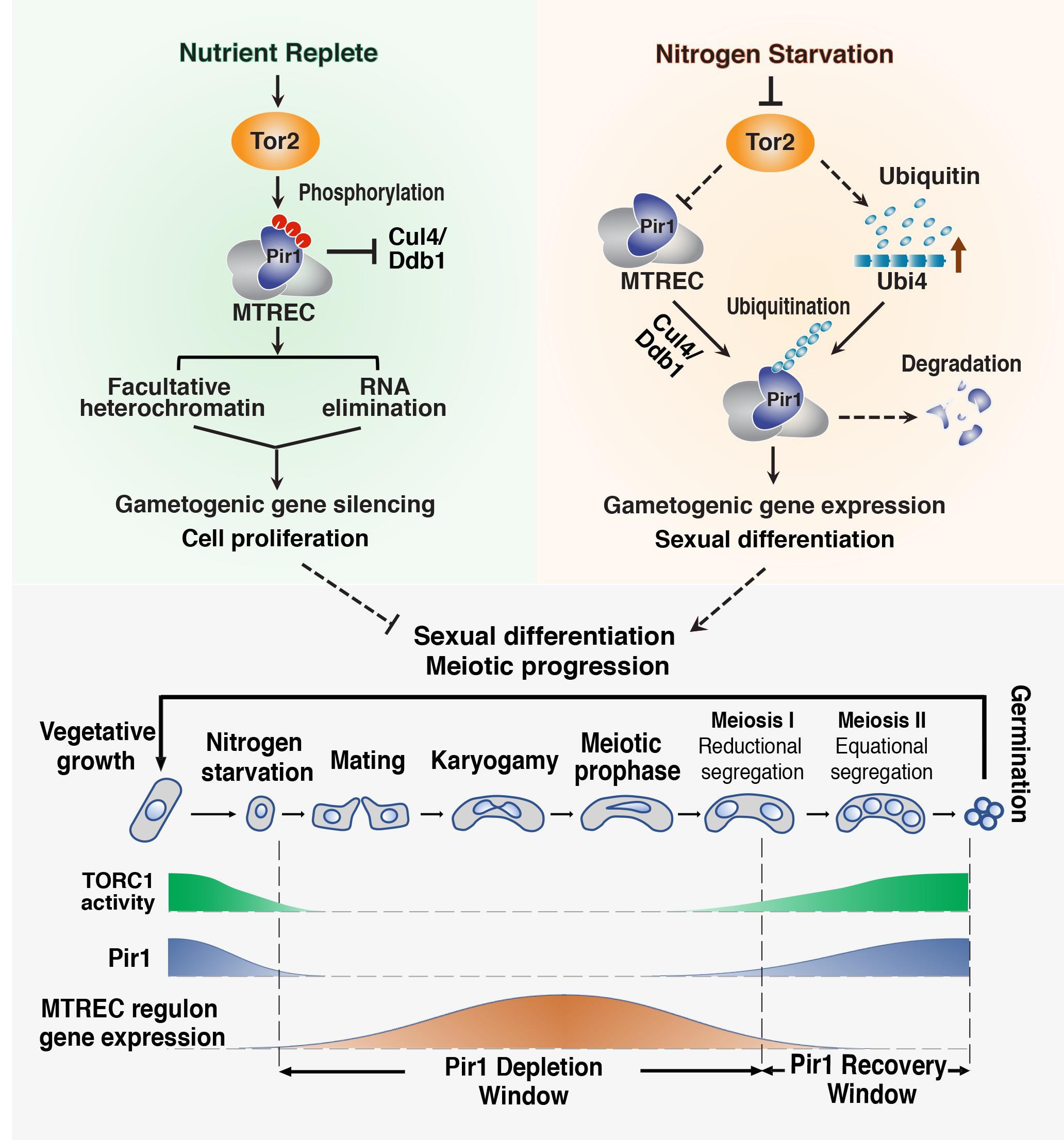
Implications of Defects in RNA Elimination and Heterochromatin Assembly: Defects that result in inappropriate gene expression can cause devastating consequences. We have found that untimely activation of the silenced gametogenic program (such as in cells lacking RNAi machinery or the RNA elimination factor Mmi1) triggers uniparental disomy (UPD), in which cells contain two copies of a chromosome originating from only one parent. UPD is frequently associated with congenital disorders and cancer. Our analyses revealed that UPD is linked to the misexpression of gametogenic genes that are only expressed in cells undergoing sexual differentiation. For example, we have reported that misexpression of Rec8, which promotes reductional chromosome segregation in cell undergoing gametogenesis, causes aberrant chromosome segregation and UPD. This work has important implications for understanding the UPD phenomenon in human populations and provides a solid foundation for the development of therapies to treat diseases, including various cancers linked to the aberrant expression of germline genes in somatic cells.
Publications
- Bibliography Link
- View Dr. Grewal's PubMed Summary.
Untimely expression of gametogenic genes in vegetative cells causes uniparental disomy
Biography

Shiv Grewal, Ph.D.
As a Distinguished Investigator at the National Cancer Institute (NCI), Dr. Grewal has dedicated his career to advancing the field of epigenetics. His groundbreaking research has significantly deepened our understanding of heterochromatin formation—a critical process that prevents improper gene expression and maintains genome stability, with important implications linked to cancer.
Dr. Grewal earned his Ph.D. at the University of Cambridge in 1992 as a Cambridge-Nehru Scholar. He began his postdoctoral fellowship at NCI, where he made the remarkable discovery that heterochromatic gene silencing is propagated epigenetically in cis, demonstrating that inheritance extends beyond just DNA. In 1998, he joined the faculty at Cold Spring Harbor Laboratory, where he identified a crucial link between genetically defined silencing proteins and histone-modifying activities. Through a series of high-impact studies, he revealed that the methylation of histone H3 at lysine 9 (H3K9me) serves as a pivotal signal for attracting essential silencing proteins, such as HP1. Moreover, he was the first to demonstrate that site-specific histone H3 methylation patterns (H3K9me and H3K4me) govern the organization of chromosomes into distinct structural and functional domains. Another groundbreaking achievement was his discovery of the connection between RNAi and heterochromatin assembly, a finding recognized as the "Breakthrough of the Year" by Science in 2002.
In 2003, Dr. Grewal returned to NCI, continuing to push the boundaries of epigenetic research. Notably, he defined core principles of epigenetic inheritance by elucidating how heterochromatin is inherited in a self-templating manner. He was first to demonstrate that heterochromatin facilitates its own reassembly through a read-write mechanism. In this process, preexisting methylated histones act as an epigenetic template, providing binding site for recruitment of histone methyltransferase ("read"), which subsequently deposits H3K9me ("write"), enabling a self-templating assembly of heterochromatin.
Dr. Grewal’s influential research, including three papers recognized as landmark discoveries by Nature, has been crucial in furthering the understanding of diseases like cancer. His significant contributions have earned him numerous prestigious awards, such as the Newcomb-Cleveland Prize, the NIH Merit Award, and the NIH Director's Award. Additionally, he has been elected to the United States National Academy of Sciences and the American Academy of Arts and Sciences.
Job Vacancies
We have no open positions in our group at this time, please check back later.
To see all available positions at CCR, take a look at our Careers page. You can also subscribe to receive CCR's latest job and training opportunities in your inbox.
Team


News

Lab Life

Grewal lab 2014

Grewal lab 2017