
Daniel W. McVicar, Ph.D.
- Center for Cancer Research
- National Cancer Institute
- Building 560, Room 21-89B
- Frederick, MD 21702-1201
- 301-846-5163
- mcvicard@mail.nih.gov
RESEARCH SUMMARY
Dr. McVicar’s laboratory studies the regulation of innate immune cell metabolism and function in cancer. His research team uses a variety of cellular and molecular techniques to understand the biology and biochemistry of a variety of receptor systems including the triggering receptors expressed on myeloid cells (TREM) that regulate neutrophils, macrophages, monocytes, dendritic cells and platelets, and the killer Ig-like receptors (KIRs) that regulate natural killer (NK) cells. Components of signaling cascades identified by the laboratory are interrogated using murine models of infection and/or inflammation-associated cancers including hepatocellular carcinoma and colorectal cancer. Dr. McVicar leads the Cancer Innovation Laboratory (CIL) in his role as CCR Deputy Director.
Areas of Expertise
Daniel W. McVicar, Ph.D.
Research
Immunometabolism in Cancer Inflammation
The tumor microenvironment represents a complex, multicellular milieu, where various cell types compete for resources necessary for their function(s). In the case of the tumor, cells must rapidly reproduce while promoting expansion of the host vasculature to maintain nutrient supplies. At the same time, tumor cells subjugate host defense mechanisms to thwart rejection. Many mechanisms for tumor-mediated immunosuppression have been described, and several involve mechanisms based on metabolic alterations. For example, highly glycolytic tumors deplete glucose in the microenvironment suppressing T cell proliferation and activation. The resulting elevated lactate from the tumor can transcriptionally reprogram tumor-associated macrophages into immunosuppressive cells promoting tumor growth and progression. My laboratory has a long history of studying the expression, signaling and function of receptors expressed by innate immune cells in the context of cancer. In recent years, interest in understanding stimulation-induced alterations in cellular metabolic processes and the critical role that they play in enabling immune cells to meet the enhanced metabolic demands associated with activation has exploded. Immunometabolism presents exciting possibilities for unraveling, and possibly therapeutically exploiting, the substantial metabolic interactions between inflammatory cells and the tumor. Therefore, my laboratory has begun to focus on the dissection of the biochemical mechanisms underlying activation-induced metabolic changes in innate immune cells.
Macrophage Metabolic Reprogramming. For example, we have been investigating the mechanisms of metabolic reprogramming in macrophages. Classical activation of macrophages with proinflammatory stimuli such as LPS and IFNg results in the generation of so called M1 macrophages that are directly bactericidal and tumoricidal, and promote adaptive immune responses. Metabolically these cells downregulate their use of oxidative phosphorylation and adapt a Warburg-like metabolism dependent on aerobic glycolysis termed “Glycolytic Commitment”. We found that autologous nitric oxide (NO) was necessary and sufficient to drive the decline in oxidative phosphorylation associated with “glycolytic commitment” and that IL-10 regulated this process. Using unbiased metabolomics, expression analysis, metabolic flux analysis, 13C carbon tracing experiments, and enzymology, we have now extensively defined the role of NO in this metabolic reprogramming. We find that NO is responsible for the rerouting of carbon usage at several points in the metabolic cascade, regulation of inflammatory mediators, and alterations of electron transport chain (ETC) components.
Niche Specific Metabolic Adaptations in Cancer. Because the tumor microenvironment represents a unique physiological niche, we have taken the approach of dissecting the metabolic interaction of leukocytes with their unique niches. We found that the peritoneal cavity is a unique metabolic niche enriched glutamine and glutamate as compared to serum and that resident peritoneal macrophages utilize these fuels to maintain high rates of mitochondrial fitness through a unique engagement of complex II of the ETC.
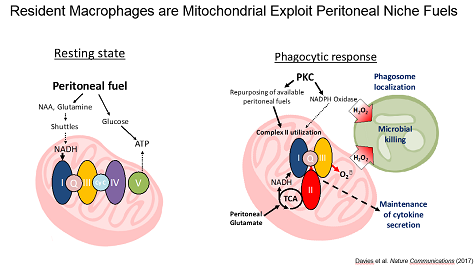
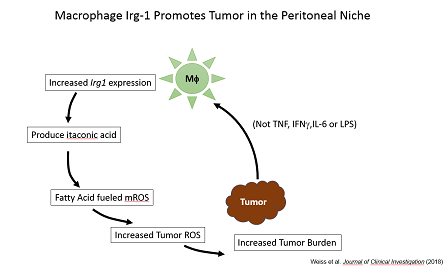
Moreover, we found that resident peritoneal macrophages (pResf) undergo unique metabolic reprogramming when tumors grow in the peritoneal cavity. These pResf promote tumor growth by expressing the metabolic enzyme Irg1.
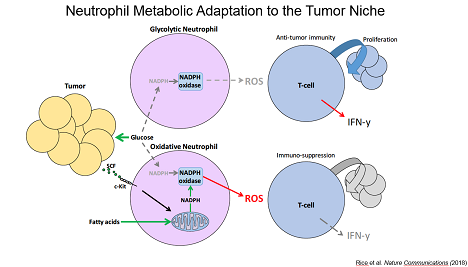
Lastly, we identified a cancer-associated population of neutrophils that upregulate mitochondrial function to generate NADPH allowing them to suppress T cell activity even when glucose utilization is limiting. Together, our data have defined unique metabolic adaptations with substantial impact on cancer inflammation.
Publications
- Bibliography Link
- View Dr. McVicar's PubMed Summary.
Nitric Oxide in Macrophage Immunometabolism: Hiding in Plain Sight
Metabolic but not transcriptional regulation by PKM2 is important for natural killer cell responses
Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase
Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression
Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors
Biography

Daniel W. McVicar, Ph.D.
Dr. McVicar obtained his Ph.D. from the Medical College of Virginia, Virginia Commonwealth University, where he studied adoptive immunotherapy of brain tumors. He joined the NCI in 1990, completed a postdoctoral fellowship studying leukocyte signaling, and served as a Senior Investigator in the Cancer and Inflammation Program. In 2016, Dr. McVicar was appointed as a Deputy Director for the Center for Cancer Research, the NCI's intramural program. He currently leads the Cancer Innovation Laboratory.
Job Vacancies
We have no open positions in our group at this time, please check back later.
To see all available positions at CCR, take a look at our Careers page. You can also subscribe to receive CCR's latest job and training opportunities in your inbox.
Team


News
Lab Life

Jonathan Weiss, Ph.D., Staff Scientist

Jeffrey Subleski, Biologist

Marieli Gonzalez-Cotto, Ph.D., Postdoctoral Fellow (CRTA)

Erika Palmieri, Ph.D. Postdoctoral Fellow (Visiting)

Christopher Rice, Ph.D., Postdoctoral Fellow (Visiting)

Luke Davies, BSc., Research Collaborator