
Deborah K. Morrison, Ph.D.
- Center for Cancer Research
- National Cancer Institute
- Advanced Technology Research Facility, D3027
- Frederick, MD 21701
- 301-846-1733
- morrisod@mail.nih.gov
RESEARCH SUMMARY
Dr. Morrison investigates the signal transduction pathways regulated by the Ras family of GTPases. These pathways are required for normal growth and development, and mutations in various pathway components can function as disease drivers in human cancer and certain developmental disorders. Dr. Morrison has extensive expertise in the identification of protein phosphorylation sites and in the proteomic analysis of signaling complexes, which has been instrumental for her studies investigating the role of the Raf protein kinases and the KSR and CNK scaffolds in Ras-dependent signaling. Her work seeks to identify critical regulatory mechanisms that may prove helpful in the diagnosis and/or treatment of human disease states with aberrant Ras-pathway signaling.
Areas of Expertise
Deborah K. Morrison, Ph.D.
Research
Regulation of Signal Transduction Pathways
The proliferative state of normal cells is precisely regulated by signals received from the extracellular environment. Understanding how these signals are relayed from the cell surface to the nucleus is the major objective of the Cellular Growth Mechanisms Section. Studies in this area are particularly relevant to cancer research because release from regulatory controls at any point along the signaling cascade can result in uncontrolled cell growth or oncogenesis. We have chosen to focus our studies on the pathway regulated by the Ras GTPase, where our aims are to further elucidate the processes involved in signal transduction and to identify critical regulatory points linked to tumor formation.
The transmission of growth and developmental signals from the membrane to the nucleus requires the coordinated action of a diverse set of proteins. Previous work from our laboratory and others has demonstrated that the Raf serine/threonine kinases serve as central intermediates in many signaling pathways, functioning downstream of activated tyrosine kinases and Ras and upstream of mitogen-activated protein kinase (MAPK) and MAPK kinase (MKK or MEK). A major goal of our research has been to elucidate the mechanisms regulating Raf activity, with a particular emphasis on understanding the role that phosphorylation events and protein interactions play in Raf regulation.
Our laboratory's research efforts have also focused on scaffold proteins involved in signal transduction, such as the kinase suppressor of Ras (KSR) family. KSR was originally identified in Drosophila and C. elegans and is an evolutionarily conserved component of the Ras pathway. Our studies examining the mammalian KSR proteins are directed towards determining the function and regulation of these scaffolds in cell signaling.
Finally, our laboratory has had an ongoing interest in using mass spectrometry to analyze signaling complexes. By taking a proteomics approach, we hope to identify new components involved in Ras-dependent signal transduction.
Publications
- Bibliography Link
- View Dr. Morrison's PubMed Summary
RASopathy mutations provide functional insight into the BRAF cysteine-rich domain and reveal the importance of autoinhibition in BRAF regulation
Structural insights into the BRAF monomer-to-dimer transition mediated by RAS binding
Development of a High-throughput NanoBRET Screening Platform to Identify Modulators of the RAS/RAF Interaction
Distinct Binding Preferences between Ras and Raf Family Members and the Impact on Oncogenic Ras Signaling
M-Ras/Shoc2 signaling modulates E-cadherin turnover and cell-cell adhesion during collective cell migration
Biography

Deborah K. Morrison, Ph.D.
b>Dr. Deborah Morrison obtained her Ph.D. from Vanderbilt University School of Medicine. She then began studying signal transduction as a postdoctoral fellow in the laboratories of Dr. Thomas Roberts at Harvard Medical School and Dr. Lewis Williams at the University of California-San Francisco. Dr. Morrison joined the ABL-Basic Research Program in 1990 and became head of the Cellular Growth Mechanisms Section in 1995. From 1996 to 1997, she was on sabbatical in the laboratory of Dr. Gerald Rubin at the University of California-Berkeley. Dr. Morrison joined the NCI Center for Cancer Research in 1999 and became Chief of the Laboratory of Cell and Developmental Signaling in 2006. In 2022, Dr. Morrison was elected into the National Academy of Sciences.
Job Vacancies
We have no open positions in our group at this time, please check back later.
To see all available positions at CCR, take a look at our Careers page. You can also subscribe to receive CCR's latest job and training opportunities in your inbox.
Team

News
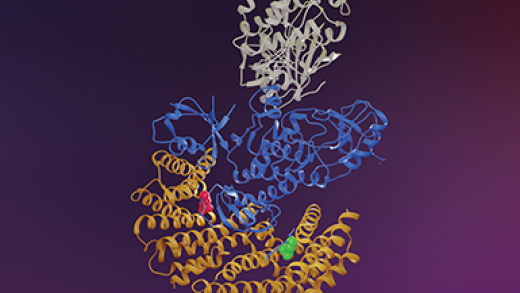

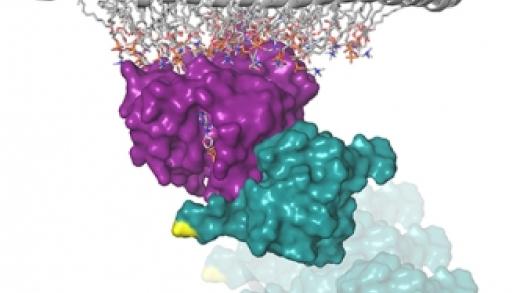
Covers

Distinct Binding Preferences between Ras and Raf Family Members and the Impact on Oncogenic Ras Signaling
About the Cover
The Ras and Raf family members are depicted as locks and keys, respectively. In this issue of Molecular Cell, Morrison et al. (pp. 872–884) suggest that C-Raf is the skeleton key that binds all Ras family members with high affinity, whereas B-Raf exhibits a striking preference for K-Ras. The hypervariable region (HVR) of K-Ras uniquely contains a polybasic region (PBR) that engages the B-Raf N-segment, allowing K-Ras to bind all Raf kinases with high affinity, which may contribute to its increased involvement in human cancer. Cover art by Joe Meyer (Scientific Publications, SAIC-Frederick).
Abstract
The Ras GTPases are frequently mutated in human cancer, and, although the Raf kinases are essential effectors of Ras signaling, the tumorigenic properties of specific Ras-Raf complexes are not well characterized. Here, we examine the ability of individual Ras and Raf proteins to interact in live cells using bioluminescence resonance energy transfer (BRET) technology. We find that C-Raf binds all mutant Ras proteins with high affinity, whereas B-Raf exhibits a striking preference for mutant K-Ras. This selectivity is mediated by the acidic, N-terminal segment of B-Raf and requires the K-Ras polybasic region for high-affinity binding. In addition, we find that C-Raf is critical for mutant H-Ras-driven signaling and that events stabilizing B-Raf/C-Raf dimerization, such as Raf inhibitor treatment or certain B-Raf mutations, can allow mutant H-Ras to engage B-Raf with increased affinity to promote tumorigenesis, thus revealing a previously unappreciated role for C-Raf in potentiating B-Raf function.
Distinct Binding Preferences between Ras and Raf Family Members and the Impact on Oncogenic Ras Signaling. Elizabeth M Terrell 1, David E Durrant 1, Daniel A Ritt 1, Nancy E Sealover 2, Erin Sheffels 2, Russell Spencer-Smith 1, Dominic Esposito 3, Yong Zhou 4, John F Hancock 4, Robert L Kortum 2, Deborah K Morrison 5. Mol. Cell, 2019 Dec 19;76(6):872-884.

Effects of Raf dimerazation and its inhibition on normal and disease-associated Raf signaling
About the Cover
The Raf kinases are essential for normal Ras-Raf-MEK-ERK pathway signaling, and activating mutations in components of this pathway are associated with human cancer and the related developmental disorders known as RASopathies. In this issue, Freeman et al. (pp. 751–758) demonstrate that Raf dimerization is required for normal Ras-dependent signaling as well as the function of certain disease-associated Raf mutants. A peptide inhibitor based on the Raf dimer interface, DI1 (superhero), was found to inhibit Raf dimerization (trains colliding) and Raf signaling. This study identifies the Raf dimer interface as a therapeutic target and has important implications for the treatment of human disease states with elevated Ras-Raf-MEK-ERK pathway signaling. Cover art by Allen Kane (Scientific Publications, SAIC-Frederick).
Abstract
Raf kinases are essential for normal Ras-Raf-MEK-ERK pathway signaling, and activating mutations in components of this pathway are associated with a variety of human cancers, as well as the related developmental disorders Noonan, LEOPARD, and cardiofaciocutaneous syndromes. Although the Raf kinases are known to dimerize during normal and disease-associated Raf signaling, the functional significance of Raf dimerization has not been fully elucidated. Here, using mutational analysis and a peptide inhibitor, we show that dimerization is required for normal Ras-dependent Raf activation and for the biological function of disease-associated Raf mutants with moderate, low, or impaired kinase activity. However, dimerization is not needed for the function of B-Raf mutants with high catalytic activity, such as V600E-B-Raf. Importantly, we find that a dimer interface peptide can effectively block Raf dimerization and inhibit Raf signaling when dimerization is required for Raf function, thus identifying the Raf dimer interface as a therapeutic target.
Effects of Raf dimerazation and its inhibition on normal and disease-associated Raf signaling. Freeman AK, Ritt DA, and Morrison DK. Mol Cell. 2013 Feb 21;49(4):751-8.

A CC-SAM, for coiled coil-sterile α motif, domain targets the scaffold KSR-1 to specific sites in the plasma membrane
About the Cover
KSR1 is a conserved component of the RAS/MAPK pathway that functions as a pseudokinase/protein scaffold. In this issue, Koveal et al week uncovers a second domain in the scaffold KSR1 named CC-SAM that mediates plasma membrane binding. The image shows the NMR solution structure of CC-SAM, highlighting the α helix required for membrane binding. Cover image from Dorothy Koveal (Department of Molecular Biology, Cell Biology and Biochemistry, Brown University)
Abstract
Kinase suppressor of Ras-1 (KSR-1) is an essential scaffolding protein that coordinates the assembly of the mitogen-activated protein kinase (MAPK) module, consisting of the MAPK kinase kinase Raf, the MAPK kinase MEK (mitogen-activated or extracellular signal-regulated protein kinase kinase), and the MAPK ERK (extracellular signal-regulated kinase) to facilitate activation of MEK and thus ERK. Although KSR-1 is targeted to the cell membrane in part by its atypical C1 domain, which binds to phospholipids, other domains may be involved. We identified another domain in KSR-1 that we termed CC-SAM, which is composed of a coiled coil (CC) and a sterile α motif (SAM). The CC-SAM domain targeted KSR-1 to specific signaling sites at the plasma membrane in growth factor-treated cells, and it bound directly to various micelles and bicelles in vitro, indicating that the CC-SAM functioned as a membrane-binding module. By combining nuclear magnetic resonance spectroscopy and experiments in cultured cells, we found that membrane binding was mediated by helix α3 of the CC motif and that mutating residues in α3 abolished targeting of KSR-1 to the plasma membrane. Thus, in addition to the atypical C1 domain, the CC-SAM domain is required to target KSR-1 to the plasma membrane.
A CC-SAM, for coiled coil-sterile α motif, domain targets the scaffold KSR-1 to specific sites in the plasma membrane. Dorothy Koveal 1, Natasha Schuh-Nuhfer, Daniel Ritt, Rebecca Page, Deborah K Morrison, Wolfgang Peti. Sci Signal. 2012 Dec 18;5(255).

The CNK1 scaffold binds cytohesins and promotes insulin pathway signaling
About the Cover
In this issue, Lim et al. (pp.1496-1506) report that the CNK1 scaffold interacts constitutively with the cytohesin family of ARF guanine nucleotide exchange factors, and facilitates the recruitment of the cytohesins to the cell surface in response to insulin treatment. Shown here is a stylized immunofluorescence (IF) image of insulin-treated cells, using antibodies directed against CNK1 (green) and cytohesin-2 (red), showing their colocalization (yellow) in the cytosol and at the plasma membrane. At the plasma membrane, the CNK1/cytohesin interaction promotes signaling from plasma membrane-bound Arf GTPases to the PIP5Ks to generate a PIP2-rich microenvironment that is critical for the membrane recruitment of IRS1 and signal transmission to the PI3K/AKT cascade. Cover art by Allen Kane (Scientific Publications, SAIC-Frederick).
Abstract
Protein scaffolds play an important role in signal transduction, regulating the localization of signaling components and mediating key protein interactions. Here, we report that the major binding partners of the Connector Enhancer of KSR 1 (CNK1) scaffold are members of the cytohesin family of Arf guanine nucleotide exchange factors, and that the CNK1/cytohesin interaction is critical for activation of the PI3K/AKT cascade downstream from insulin and insulin-like growth factor 1 (IGF-1) receptors. We identified a domain located in the C-terminal region of CNK1 that interacts constitutively with the coiled-coil domain of the cytohesins, and found that CNK1 facilitates the membrane recruitment of cytohesin-2 following insulin stimulation. Moreover, through protein depletion and rescue experiments, we found that the CNK1/cytohesin interaction promotes signaling from plasma membrane-bound Arf GTPases to the phosphatidylinositol 4-phosphate 5-kinases (PIP5Ks) to generate a PIP(2)-rich microenvironment that is critical for the membrane recruitment of insulin receptor substrate 1 (IRS1) and signal transmission to the PI3K/AKT cascade. These findings identify CNK1 as a new positive regulator of insulin signaling.
The CNK1 scaffold binds cytohesins and promotes insulin pathway signaling. Junghwa Lim 1, Ming Zhou, Timothy D Veenstra, Deborah K Morrison. Genes Dev. 2010 Jul 15;24(14):1496-506.

Caspase-dependent cleavage disrupts the ERK cascade scaffolding function of KSR1
About the Cover
KSR1 is a protein scaffolding that facilitates ERK activation at the plasma membrane, as depicted by the colocalization of KSR1 and ERK at the cell surface (yellow in the merged image). In this issue, McKay and Morrison (pp. 26225-26234) demonstrate that caspase-dependent cleavage KSR1 in apoptotic cells disrupts the ability of KSR1 to promote ERK activation and ERK survival signaling. Pyo-tagged KSR1 staining is green, ERK staining is red, and yellow represents colocalization of the two proteins. Cover image from Melissa McKay (NCI-Frederick).
Abstract
Kinase suppressor of Ras 1 (KSR1) is a protein scaffold that facilitates ERK cascade activation at the plasma membrane, a critical step in the signal transduction process that allows cells to respond to survival, proliferative, and differentiative cues. Here, we report that KSR1 undergoes caspase-dependent cleavage in apoptotic cells and that cleavage destroys the scaffolding function of the full-length KSR1 protein and generates a stable C-terminal fragment that can inhibit ERK activation. KSR1 is cleaved in response to multiple apoptotic stimuli and occurs in vivo during the involution of mouse mammary tissues, a morphogenic process requiring cellular apoptosis. In addition, we find that in comparison with KSR1(-/-) mouse embryonic fibroblasts expressing wild type KSR1 (WT-KSR1), cells expressing a cleavage-resistant KSR1 protein (DEVA-KSR1) exhibit reduced apoptotic signaling in response to tumor necrosis factor-alpha/cycloheximide treatment. The effect of DEVA-KSR1 expression was found to correlate with increased levels of active phosphoERK and could be significantly reversed by treating cells with the MEK inhibitor U0126. In contrast, reduced phosphoERK levels and enhanced apoptotic signaling were observed in cells constitutively expressing the C-terminal KSR1 fragment (CTF-KSR1). Moreover, we find that cleavage of WT-KSR1 correlates with a dramatic reduction in active phosphoERK levels. These findings identify KSR1 as a caspase target and suggest that cleavage of the KSR1 scaffold represents another mechanism whereby caspases down-regulate ERK survival signaling to promote cellular apoptosis.
Caspase-dependent cleavage disrupts the ERK cascade scaffolding function of KSR1. Melissa M McKay 1, Deborah K Morrison. J Biol Chem. 2007 Sep 7;282(36):26225-34.