
Stephen H. Hughes, Ph.D.
- Center for Cancer Research
- National Cancer Institute
- Building 535, Room 303D
- Frederick, MD 21702-1201
- 301-846-1619
- hughesst@mail.nih.gov
RESEARCH SUMMARY
Dr. Hughes is internationally renowned for his work on the roles of reverse transcriptase (RT) and integrase (IN). He is interested in how HIV becomes resistant to RT and IN inhibitors, in developing drugs that are more effective against the known resistance mutations, and in how the clonal expansion of HIV-infected cells contributes to the formation and persistence of the reservoir that has made it impossible to cure HIV infections with the available drugs. In his role as NIH Scientist Emeritus, Dr. Hughes interacts with scientists inside and outside of the HIV-DRP, providing advice and mentoring.
Areas of Expertise
Research
HIV-1 Reverse Transcriptase and Integrase
Dr. Hughes has a long-standing interest in two viral enzymes that are essential for HIV replication: reverse transcriptase (RT) and integrase (IN). Although much of his work focused on understanding the mechanisms that underlie drug resistance, and using that information to develop more effective anti-RT and anti-IN drugs, his laboratory did experiments that furthered our understanding of the roles that these enzymes play in viral replication. In addition, they used analysis of HIV integration sites to better understand chromatin structure and function and as a tool to study the nature and origin of the HIV reservoir in patients on antiretroviral therapy.
Background. HIV-1 is the causative agent of AIDS. The three viral enzymes — RT, IN, and protease (PR) — have essential roles in the replication of HIV-1 and are the targets for the most potent anti-HIV drugs. Considerable progress has been made in treating HIV-infected patients with three- and four-drug regimens and there has been good progress in the development of drug treatments that can reduce or prevent new infections. A potent preventive vaccine would be ideal; however, despite a huge effort, the goal of developing an effective vaccine remains elusive. In the absence of an effective vaccine, reducing the transmission of HIV-1 must rely on barrier methods and/or drug treatments. There are two ways that anti-HIV drugs can be used to reduce viral transmission: (1) effective therapy in infected patients can reduce the viral load, making it less likely that an infected individual will transmit the virus to a partner; and (2) treating the uninfected partner with an anti-HIV drug can block transmission. Because most new infections are caused by a single virus, blocking transmission is an attractive option and there is now good evidence that giving an anti-HIV drug to the uninfected partner can significantly reduce viral transmission if the uninfected partner is compliant. Because of the problem of drug resistance, it would be better to use drugs with nonoverlapping resistance profiles for treatment and prophylaxis. Treatment would have to be long term and, for this reason, drug toxicity is an important consideration, which argues against the use of nucleoside RT inhibitors (NRTIs). It would also be better to block infection before the viral DNA is integrated, which argues against the use of PR inhibitors. The two remaining options among the major classes of anti-HIV drugs are nonnucleoside RT inhibitors (NNRTIs) and IN strand-transfer inhibitors (INSTIs). In addition to their work on NRTIs and the mechanisms of NRTI resistance, Dr. Hughes and his colleagues and collaborators used a combined approach that involved structural analysis, biochemistry, virology, modeling, toxicity testing, and chemistry to design, synthesize, and evaluate new NNRTIs and INSTIs. They made good progress in developing new compounds that are effective against the wild-type (WT) and common drug-resistant viruses and that have good therapeutic indexes in tests done in cultured cells.
HIV-1 RT. There are two clinically important classes of inhibitors of HIV-1 RT: NRTIs and NNRTIs. A major early focus of Dr. Hughes’ work on HIV-1 RT was on the mechanisms of inhibitor resistance. There is now a reasonably good understanding of the mechanism(s) of NNRTI resistance, and considerable progress has been made in understanding NRTI resistance. Dr. Hughes studied the mechanisms of resistance to nucleoside analogs (NRTIs) in collaboration with Dr. Arnold (Rutgers University) and elucidated the mechanism of action of compounds that do not cause an immediate or complete block to DNA synthesis. In addition, Dr. Hughes worked on the development of NNRTIs. Work with chemistry collaborators, Drs. Joel Schneider, Gary Pauly, and Craig Thomas, led to the development of compounds that are able to inhibit, in a one-round replication assay, WT and several common NNRTI-resistant viruses with IC50s below 5 nM. In cultured cells, these compounds have CC50s that are more than 4 logs higher than their IC50s.
Almost all of the work that has been done on the mechanisms that underlie drug resistance in HIV-1 RT has been done with RTs from subgroup B viruses. Although subgroup B is the most common strain in the United States, it is not the major subgroup worldwide. Dr. Hughes’ laboratory showed that the RT of a subgroup C virus behaved quite similarly to a standard subgroup B RT, both in terms of its biochemical properties and the effects of a limited set of resistance mutations.
HIV-1 IN. Like RT, HIV-1 IN is an important drug target; however, as is the case for all anti-HIV drugs, treatment with INSTIs leads to resistance. Dr. Hughes and his colleagues and collaborators made good progress on two fronts: understanding how mutations in IN confer resistance to the currently available compounds and developing new INSTIs that are effective against the common drug-resistance mutations. Dr. Terrence Burke and his colleagues synthesized novel INSTIs that the Hughes laboratory tested for their effects on viral replication and their toxicity in cultured cells.
Dr. Craigie’s laboratory makes large amounts of purified recombinant HIV-1 IN (both WT and mutant). Dr. Lyumkis’s laboratory uses the purified IN to obtain, by cryo-electron microscopy, high-resolution structures of HIV-1 IN in complexes with the ends of the viral DNA and INSTIs. These structures were solved at a sufficiently high resolution to show ordered waters and the structures are being used in drug design and drug development. Dr. Burke has synthesized several novel compounds that have IC50s in the low nanomolar range in a one-round replication assay. The best compounds effectively inhibit both WT IN and most of the common drug-resistant variants. Based on tests done in cultured cells, these compounds have excellent therapeutic indexes (the CC50s are more than 3 logs higher than the IC50s). With support from the NCI, the most promising of the new compounds is being tested by Dr. Jeffrey Lifson (Leidos) for uptake, stability, and toxicity in macaques.
Dr. Hughes worked with Drs. Xiaolin Wu, Frank Maldarelli, John Coffin, John Mellors, and Mary Kearney to determine the distribution of integration sites in samples obtained from HIV-infected donors. Dr. Hughes and his colleagues showed that there is extensive clonal expansion of HIV-infected cells in patients. The initial distribution of HIV proviruses, which is reasonably well understood, is determined by interactions of the HIV pre-integration complex with host factors (primarily CPSF6 and LEDGF) and chromatin. HIV preferentially integrates into the bodies of highly expressed genes. They showed that, in people living with HIV, the initial distribution of the proviruses is affected by selections for and against cells that have proviruses integrated in a subset of genes. The primary negative selection is against cells that have proviruses integrated in highly expressed genes; that selection is stronger for cells in which the proviruses are integrated in the same orientation as the gene. This negative selection appears to depend on the provirus interfering with the expression of the host gene, which is why the selection is stronger in highly expressed genes. There is a positive selection for cells with a provirus integrated in particular introns of seven oncogenes: BACH2, STAT5B, MKL2, MKL1, IL2RB, MYB, and POU2F1. Despite the fact that the primary mechanism of oncogene activation appears to be promoter insertion, these clonally expanded cells are not transformed. However, oncogene activation accounts for only a small fraction (2-3%) of the clones of HIV-infected cells. It appears that the primary forces that drive the clonal expansion of HIV-infected T cells are the same as the forces that drive the clonal expansion of uninfected T cells: antigen and cytokine stimulation.
Dr. Hughes and his colleagues showed that clones of infected cells arise in the first few weeks after a person is infected with HIV. Some of the clones that arise early can persist for years. Because there are some experiments that cannot be performed with samples obtained from human donors, Dr. Hughes collaborated with Dr. Jeffrey Lifson to develop a system to study the behavior of simian immunodeficiency virus (SIV)-infected cells in macaques. They showed that the overall distribution of SIV proviruses/integration sites in macaques is very similar to the overall distribution of HIV proviruses/integration sites in human donors. They also showed that there is extensive clonal expansion of SIV-infected cells in macaques, and that the infected cells behave in ways that are quite similar to the behavior of HIV-infected cells in humans.
Initially there was a question of whether any of the clonally expanded cells in HIV infected individuals on successful ART carried an infectious provirus. Dr. Hughes and his colleagues showed not only that there are highly expanded clones of cells that carry infectious proviruses, but that some cells in these clones can release detectable amounts of virus into the blood. Importantly, even after periods of more than nine years, large clones that carry infectious proviruses can grow larger or smaller in individuals on effective ART.
In experiments done with samples from donors on successful long-term ART, Dr. Hughes, working with Drs. Xiaolin Wu, John Mellors, and Danny Douek (NIAID) showed that clones of HIV-infected CD4+ T cells are much more stable than are clones of uninfected CD4+T cells. Because successful ART blocks new HIV infections, all of the infected clones must have derived from T cells that were infected before ART was initiated. Thus, in those on successful long-term ART, all the infected clones are “old”. In order for a clone to survive long enough to become old, it would have had to be relatively stable. In contrast the clones of infected T cells could have arisen either before or after ART was initiated (which means that the uninfected clones are a mixture of young and old).
Insertional mutagenesis by retroviruses is a common cause of oncogenesis in animals. In the animal models, integration of a provirus in or near an oncogene can enhance the expression of the oncogene, affect the nature of the expressed oncogene protein, or both. It has been proposed that some retroviruses, including HIV, do not cause cancers by insertional mutagenesis. As has already been discussed, Dr. Hughes and his colleagues showed that integration of an HIV provirus in seven oncogenes can lead to clonal expansion of the infected cells without causing the cells to be transformed. Chronic HIV infection is known to increase the risk of certain type of cancers, including Hodgkin’s lymphoma, non-Hodgkin’s lymphoma, Kaposi’s sarcoma, and HPV-associated cervical cancers. It is likely that much of the reported increase in cancer incidence in HIV-positive individuals is the result of HIV-induced immunodeficiency, which allows greater replication of other viruses that are known to cause human cancers, including KSHV, EBV, and HPV. As would be expected if these viruses are the primary cause of many of the cancers that arise in people living with HIV, much of the increase in cancer incidence in HIV-positive individuals involves cell types that are not readily infected by HIV.
If HIV does cause cancer by insertional mutagenesis, it is reasonable to expect that HIV would efficiently infect the cancer progenitor cell. T cell lymphomas are rare, both in HIV-negative people, and in those who are HIV-positive, although it has been reported that HIV infection causes an increase in the incidence of T cell lymphomas. Drs. Hughes, Wu, and Mellors obtained samples of T cell malignancies from HIV-positive individuals and showed that HIV proviruses integrated in the STAT3 and LCK genes can play a direct role in the development of T cell lymphomas. The data suggest that the development of the T cell lymphomas is a multi-step process which helps to explain why T cell lymphomas are rare in HIV-infected individuals.
RCAS System
Mary Kearney, Stephen Hughes, and members of the Vector Design and Replication Section (VDRS) developed the RCAS System of retroviral vectors and have widely distributed these vectors to investigators for adaptation to their research programs. Learn more
Publications
- Bibliography Link
- View Dr. Hughes's PubMed Summary
- View Dr. Hughes's ORCID Bibliography
Specific HIV integration sites are linked to clonal expansion and persistence of infected cells
Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration
Selective excision of AZTMP by drug-resistant human immunodeficiency virus reverse transcriptase
A system for tissue-specific gene targeting: transgenic mice susceptible to subgroup A avian leukosis virus-based retroviral vectors
Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 Å resolution shows bent DNA
Biography

Stephen H. Hughes, Ph.D.
Dr. Stephen H. Hughes received his Ph.D. from Harvard University under the direction of Dr. Mario Capecchi and did postdoctoral research under the direction of Drs. J. Michael Bishop and Harold Varmus at the University of California, San Francisco. From 1979 to 1984, Dr. Hughes was a Senior Staff Investigator at Cold Spring Harbor Laboratory (CSHL). In 1984 he established the Gene Expression in Eukaryotes Section in the ABL-Basic Research Program at NCI-Frederick. He became Deputy Director of the ABL-Basic Research Program in 1988 and Director of the Molecular Basis of Carcinogenesis Laboratory in 1995. In 1999 Dr. Hughes joined the HIV Drug Resistance Program (HIV DRP, renamed HIV Dynamics and Replication Program in 2015) in the National Cancer Institute and served as Chief of the Retroviral Replication Laboratory from 1999 to 2021. He was appointed Acting Director of the HIV DRP in 2005 and served as Director of the HIV DRP from 2006-2015 and Acting Chief of the Host-Virus Interaction Branch from 2005 to 2021. Dr. Hughes is a Co-Organizer of the Annual HIV DRP Conference and the David Derse Memorial Lecture and Award and has served as a Co-Organizer of the CSHL Retroviruses and Viral Vectors Meetings, the Annual Meeting on Oncogenes, and the Keystone Symposium on Pathogenesis and Control of Emerging Infections and Drug Resistant Organisms. He was named one of the most frequently cited AIDS researchers by Science Watch in 1996. In 2017 Dr. Hughes was a keynote speaker at the CSHL Retroviruses Meeting and received the 2017 Distinguished Research Career Award from The Ohio State University Center for Retrovirus Research in recognition of his substantial body of work on retroviruses. . In 2022, Dr, Hughes was a Co-Organizer of the “Fifty years of Reverse Transcriptase” meeting at Cold Spring Harbor Laboratory.
Dr. Hughes retired in 2022 and is now an NIH Scientist Emeritus.
News
Large HIV Integration Site Datasets Provide Insights into the Stability of Clones of Infected cells and the Persistence of the Reservoir
Although combination antiretroviral therapy (ART) blocks HIV replication, it is not curative because infected CD4+ T cells that carry intact, infectious proviruses persist. Understanding the behavior of clones of infected T cells is important for understanding the stability of the reservoir; however, the stabilities of clones of infected T cells in persons on long-term ART are not well defined. In a paper that was recently published in PLoS Pathogens (HIV infected CD4+ T cell clones are more stable than uninfected clones during long-term antiretroviral therapy. Guo S, Luke BT, Henry AR, Darko S, Brandt LD, Su L, Sun D, Wells D, Joseph KW, Demirov D, Halvas EK, Douek DC, Wu X, Mellors JW, Hughes SH. PLoS Pathog. 2022 Aug 31;18(8):e1010726. doi: 10.1371/journal.ppat.1010726. eCollection 2022 Aug. PMID: 36044447), Dr. Hughes and his colleagues and collaborators determined the relative stabilities of clones of infected and uninfected CD4+ T cells over time intervals of one to four years in three individuals who had been on ART for 9-19 years. Although the number of individuals in this study was relatively small, more than 19,000 integration sites were identified from each of the donors. The large integration site datasets made it possible to accurately compare the sizes of the T cell clones over time.
There are several important conclusions. First, the largest clones of uninfected T cells were larger than the largest clones of infected T cells. There are at least two explanations for this result. First, only a small fraction of the CD4+ T cells are infected. CD4+ T cells can be infected either before or after they have expanded to form a clone. If T cells are infected before they begin to expand, the cells that are infected are unlikely, based on simple probability, to be one of the rare T cells that is destined to become one of the largest T cell clones. Alternatively, if the initial infection happens in a T cell that is part of a clone that has already begun to expand, only a small fraction of the cells in the clone will be infected. Either way, the largest clones of infected cells will be smaller in size than the largest uninfected clones.
Second, clones of infected CD4+ T cells are more stable than clones of uninfected CD4+ T cells of a similar size. Because successful ART blocks new HIV infections, all of the infected clones must have derived from T cells that were infected before ART was initiated. Thus, all the infected clones in those on successful long-term ART are “old”. In order for a clone to survive long enough to become old, it would have had to be relatively stable. In contrast, the clones of infected T cells could have arisen either before or after ART was initiated (which means that the uninfected clones are a mixture of young and old).
Finally, because it is the clones that carry infectious proviruses that contribute to the reservoir, the behavior and stability of these clones are particularly important. Thus far, we only have data on the stability of three large clones of cells that carry infectious proviruses and more data are needed. One of the T cell clones that carried an infectious provirus decreased in size over time. However, in another donor, a large clone that carried an infectious provirus increased in size, becoming, at the last time point, the largest clone of infected cells in that donor. In a donor who had been on therapy for 19 years, a clone that carried an infectious provirus first increased in size and then decreased over about a four-year period. The data show that, although all three of the clones must all have persisted for at least 9 years, “old” clones that carry intact infectious proviruses can still change in size. Importantly, clones of T cell that carry infectious proviruses can still increase in size, perhaps in response to antigenic or homeostatic stimulation, many years after the cell that gave rise to the clone was first infected.
Alumni
Covers
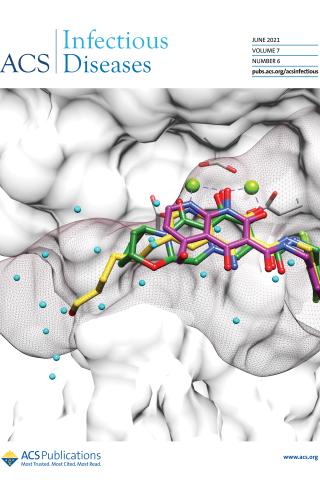
HIV-1 Integrase Inhibitors with Modifications That Affect Their Potencies against Drug Resistant Integrase Mutants
Abstract:
Integrase strand transfer inhibitors (INSTIs) block the integration step of the retroviral lifecycle and are first-line drugs used for the treatment of HIV-1/AIDS. INSTIs have a polycyclic core with heteroatom triads, chelate the metal ions at the active site, and have a halobenzyl group that interacts with viral DNA attached to the core by a flexible linker. The most broadly effective INSTIs inhibit both wild-type (WT) integrase (IN) and a variety of well-known mutants. However, because there are mutations that reduce the potency of all of the available INSTIs, new and better compounds are needed. Models based on recent structures of HIV-1 and red-capped mangabey SIV INs suggest modifications in the INSTI structures that could enhance interactions with the 3'-terminal adenosine of the viral DNA, which could improve performance against INSTI resistant mutants. We designed and tested a series of INSTIs having modifications to their naphthyridine scaffold. One of the new compounds retained good potency against an expanded panel of HIV-1 IN mutants that we tested. Our results suggest the possibility of designing inhibitors that combine the best features of the existing compounds, which could provide additional efficacy against known HIV-1 IN mutants.
On the cover:
INSTIs bound to HIV-1 IN. Compound 5j (magenta) was docked onto the structures of 4d (yellow) and BIC (green) bound to the active site of HIV-1 IN; the surface of IN is shown in white. The surface envelope of the unprocessed 3′ end of the vDNA is shown as brown mesh. Ordered water molecules bound to the active site of IN in the absence of a bound INSTI are shown in cyan.

Structure-Guided Optimization of HIV Integrase Strand Transfer Inhibitors
Abstract:
Integrase mutations can reduce the effectiveness of the first-generation FDA-approved integrase strand transfer inhibitors (INSTIs), raltegravir (RAL) and elvitegravir (EVG). The second-generation agent, dolutegravir (DTG), has enjoyed considerable clinical success; however, resistance-causing mutations that diminish the efficacy of DTG have appeared. Our current findings support and extend the substrate envelope concept that broadly effective INSTIs can be designed by filling the envelope defined by the DNA substrates. Previously, we explored 1-hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamides as an INSTI scaffold, making a limited set of derivatives, and concluded that broadly effective INSTIs can be developed using this scaffold. Herein, we report an extended investigation of 6-substituents as well the first examples of 7-substituted analogues of this scaffold. While 7-substituents are not well-tolerated, we have identified novel substituents at the 6-position that are highly effective, with the best compound (6p) retaining better efficacy against a broad panel of known INSTI resistant mutants than any analogues we have previously described.
On the cover:
Shown is an HIV integrase strand transfer inhibitor possessing a good mutant profile, bound at the catalytic site of the prototype foamy virus intasome including metal cofactors and viral DNA.
Cover design by Joseph Myer

HIV‑1 Integrase Strand Transfer Inhibitors with Reduced Susceptibility to Drug Resistant Mutant Integrases
Abstract:
HIV integrase (IN) strand transfer inhibitors (INSTIs) are among the newest anti-AIDS drugs; however, mutant forms of IN can confer resistance. We developed noncytotoxic naphthyridine-containing INSTIs that retain low nanomolar IC50 values against HIV-1 variants harboring all of the major INSTI-resistant mutations. We found by analyzing crystal structures of inhibitors bound to the IN from the prototype foamy virus (PFV) that the most successful inhibitors show striking mimicry of the bound viral DNA prior to 3'-processing and the bound host DNA prior to strand transfer. Using this concept of "bi-substrate mimicry," we developed a new broadly effective inhibitor that not only mimics aspects of both the bound target and viral DNA but also more completely fills the space they would normally occupy. Maximizing shape complementarity and recapitulating structural components encompassing both of the IN DNA substrates could serve as a guiding principle for the development of new INSTIs.
On the cover:
Mutant forms of HIV-1 IN reduce the therapeutic effectiveness of integrase strand transfer inhibitors (INSTIs). The cover figure shows the IN of prototype foamy virus complexed to a novel INSTI (gold) that retains potency against resistant mutants of HIV-1 IN. Overlain are the host and viral DNA substrates (blue and green, respectively), showing substrate mimicry by the inhibitor.
Cover design by Joseph Myer