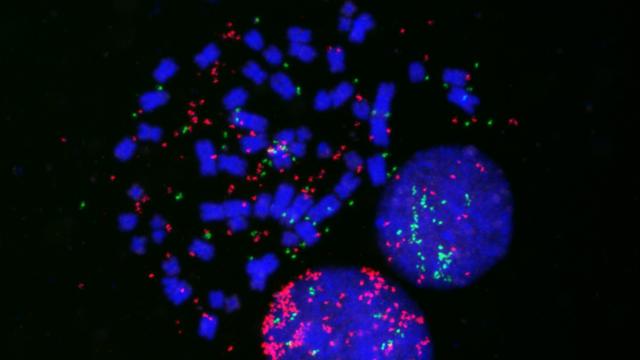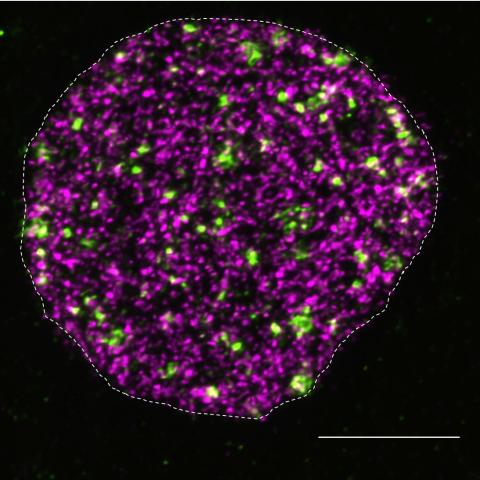
Detection of DNA breakage (green) and DNA synthesis (magenta) in the cell nucleus reveal a novel cellular mechanism that prevents the initiation of DNA replication at chromatin loops adjacent to double-stranded DNA breaks, without affecting global DNA synthesis.
CCR researchers have discovered a cellular mechanism that stops damaged DNA from replicating without affecting replication at other locations on the genome. This mechanism could be used to develop therapies to target cancer. The results were published February 19, 2025, in Nature.
Every time a cell divides, its DNA is copied, ensuring that the DNA is passed on to each new cell that is created. The replication of the genome that occurs as this happens is a complex process in which a variety of factors could go wrong and result in replication that can stall or slow. Such stalling or slowing, called replication stress, can lead to instability in the genome and is common in cancer.
The team, led by Mirit I. Aladjem, Ph.D., Senior Investigator and Deputy Chief in the Developmental Therapeutics Branch, and Research Fellow Robin Sebastian, Ph.D., discovered a previously unknown recovery process that is activated when a double-strand break occurs in DNA, meaning both strands of the DNA double helix are broken. Double-strand breaks are considered the most harmful form of DNA damage, and they can be caused both by errors in the replication process as well as by external forces such as exposure to radiation or certain chemicals. Normally, double-strand breaks would trigger cell death, but cancer cells often proliferate despite harboring numerous breaks within their genome.
The researchers conducted their study in the lab by using cells genetically engineered to deliberately cause double-strand DNA breaks at specific sites. They then used fluorescent markers and imaging methods to study how DNA synthesis was affected. In a separate procedure, they used irradiation to cause double-strand breaks and studied the results in the same manner.
“We thought that in places where there was broken DNA, DNA synthesis will stop there. But that's not what happened,” Aladjem said.
Instead, what they saw was that DNA replication was halted in extended regions in the genome that straddle the DNA break, called topologically associating domains (TADs). These TADs are kept separate from each other by a protein complex called cohesin.
Specifically, the researchers found that after a double-strand break, cohesin keeps that break-containing TAD separate from the rest of the genome. Then, two proteins, TIMELESS and TIPIN, are dislodged from their locations near double-strand breaks to inhibit DNA replication. This process isolates the affected section of DNA, allows time for the break to be repaired, and then allows the cells to continue to replicate.
To confirm this, the researchers studied double-strand breaks in cells with TIMELESS and TIPIN depleted and found that the rate of DNA replication increased and DNA synthesis continued into the TADs that contained double-strand breaks. Additionally, in cells with cohesin depleted, replication was not selectively inhibited in DNA sections that experienced a double-strand breaks.
“Basically, those proteins protect the DNA from replicating when it could have negative impacts,” Aladjem said.
Many anti-cancer treatments cause double-strand DNA breaks, and yet cancer cells continue to proliferate. If scientists are able to discover a way to block this recovery process, it could lead to irreparable damage that could selectively kill cancer cells.