
Andres M. Lebensohn, Ph.D.
- Center for Cancer Research
- National Cancer Institute
- Building 37, Room 2056C
- Bethesda, MD 20892-4256
- 240-760-7799
- andres.lebensohn@nih.gov
RESEARCH SUMMARY
Mechanisms of WNT Signaling in Development, Stem Cells and Cancer
We are interested in understanding how a relatively small number of cell signaling pathways can orchestrate the thousands of cellular events that give rise to complex organisms during embryonic development, and that maintain tissues and organs in adults. To answer this question, we focus on the WNT pathway, a fundamental signaling system that controls patterning and morphogenesis, promotes tissue renewal and regeneration, and can be a potent cancer driver when it becomes dysregulated. We use powerful genetic screens in human cells to discover new regulatory mechanisms and probe their molecular underpinnings through biochemistry and cell biology. We use organoids and mouse models to understand how these new regulatory mechanisms enable the WNT pathway to generate distinct physiological outcomes during embryonic development and tissue homeostasis. We study how dysregulation of WNT signaling causes cancer.
Areas of Expertise
Andres M. Lebensohn, Ph.D.
Research
Overview:
During embryonic development, secreted cues are received by cells and decoded through cascades of biochemical reactions called cell signaling pathways to faithfully execute many cellular programs, including self-renewal, differentiation, migration and apoptosis. Similar cell-to-cell communication mechanisms are used by stem cells to maintain and regenerate most tissues of adult organisms. Remarkably, only a dozen or so signaling pathways orchestrate the thousands of instructions required to transform a fertilized egg into a human being. This is possible because signaling pathways have evolved additional regulatory mechanisms superimposed on conserved ‘core modules’ that endow them with new biological functions.
The long-term goal of our research is to understand how signaling pathways drive the many different cellular processes that give rise to tissues and organs during embryonic development and maintain them in adult life. We seek to discover new regulatory mechanisms and understand how they modify signaling outcomes in distinct physiological contexts. Since these same signaling pathways can cause devastating diseases when they become dysregulated, understanding their context-specific regulation will also open new therapeutic avenues.
We have begun to address these questions by studying WNT signaling, a fundamental pathway that encodes positional information in animals, orchestrates patterning and morphogenesis during embryonic development, and promotes tissue renewal and regeneration in adults. Dysregulation of WNT signaling has been implicated in many diseases, including many types of cancer. Despite intensive research efforts, there are still no FDA-approved drugs that can selectively treat WNT-driven tumors, which constitute over 90% of all colorectal cancers and a large percentage of many other cancer types.
Our research strategy begins with the identification of new WNT pathway components through powerful genetic screens in cultured haploid human cells (Lebensohn et al., eLife 2016; Patel, Lebensohn et al., PLoS One 2019). We use a combination of CRISPR-based genetic analysis, cell biology and biochemistry to dissect new signaling mechanisms (Lebensohn & Rohatgi, eLife 2018; Dubey et al., eLife 2020, McKenna et al. BioRxiv 2024). We explore these mechanisms in human cells, organoid cultures and animal models to elucidate how regulatory additions to the WNT pathway give rise to distinct physiological outcomes, and how defects in these processes cause disease.
Research Projects:
New regulatory mechanisms in WNT signaling:
Through forward genetics screens in haploid human cells harboring a fluorescent reporter of WNT signaling, we discovered regulators of the intact pathway as well as those selectively required to sustain hyperactive, oncogenic signaling. These studies comprised seven genome-wide screens systematically interrogating the pathway, including screens for positive, negative and attenuating regulators of ligand-induced signaling, as well as suppressor screens following disruption of key regulators commonly mutated in WNT-driven tumors, such as the tumor suppressor APC. A comparative analysis of the screens revealed new regulatory mechanisms for ligand reception, signal transduction and transcriptional activation, as well as new gene regulatory elements. The following research projects are aimed at elucidating some of these new regulatory mechanisms and evaluating their therapeutic potential.
1) The transcription factor TFAP4 is a new limiting component of the WNT signaling pathway.
Our genetic screens uncovered a basic helix-loop-helix leucine zipper transcription factor called TFAP4 as a potential regulator of WNT signaling. We have shown that TFAP4 is a limiting component for WNT signaling activity. Furthermore, excess TFAP4 can promote ectopic activation of WNT signaling during Xenopus laevis development, causing the formation of a secondary body axis. TFAP4 is upregulated in colorectal cancer, where it mediates epithelial-mesenchymal transition (EMT) and metastasis, and its down-regulation in gastric cancer cells inhibits proliferation, induces cell cycle arrest and promotes apoptosis. We are dissecting the molecular mechanism by which TFAP4 regulates WNT signaling, and evaluating its potential as a therapeutic target.
2) A new function of the b-catenin destruction complex regulating WNT signaling through the ubiquitin ligase HUWE1.
HUWE1 is a HECT-domain ubiquitin ligase involved in dozens of cellular processes through the ubiquitination of diverse substrates. HUWE1 has been postulated as a negative regulator of WNT signaling through at least two distinct mechanisms. However, in our unbiased forward genetic screen for mediators of hyperactive WNT signaling induced by loss of the b-catenin destruction complex kinase casein kinase 1a, we identified HUWE1 as a positive regulator of the WNT pathway. We also demonstrated that HUWE1 potentiates WNT signaling in cells and in Xenopus laevis embryos. We have now found that HUWE1 promotes WNT/b-catenin signaling through a mechanism independent of the control of b-catenin protein stability. Potentiation of WNT signaling by HUWE1 requires its ubiquitin ligase activity and a subset of b-catenin destruction complex components. These results reveal a new role for some destruction complex components in mediating WNT signaling through HUWE1, distinct from their established activity in controlling b-catenin stability.
New regulatory mechanisms in R-spondin signaling:
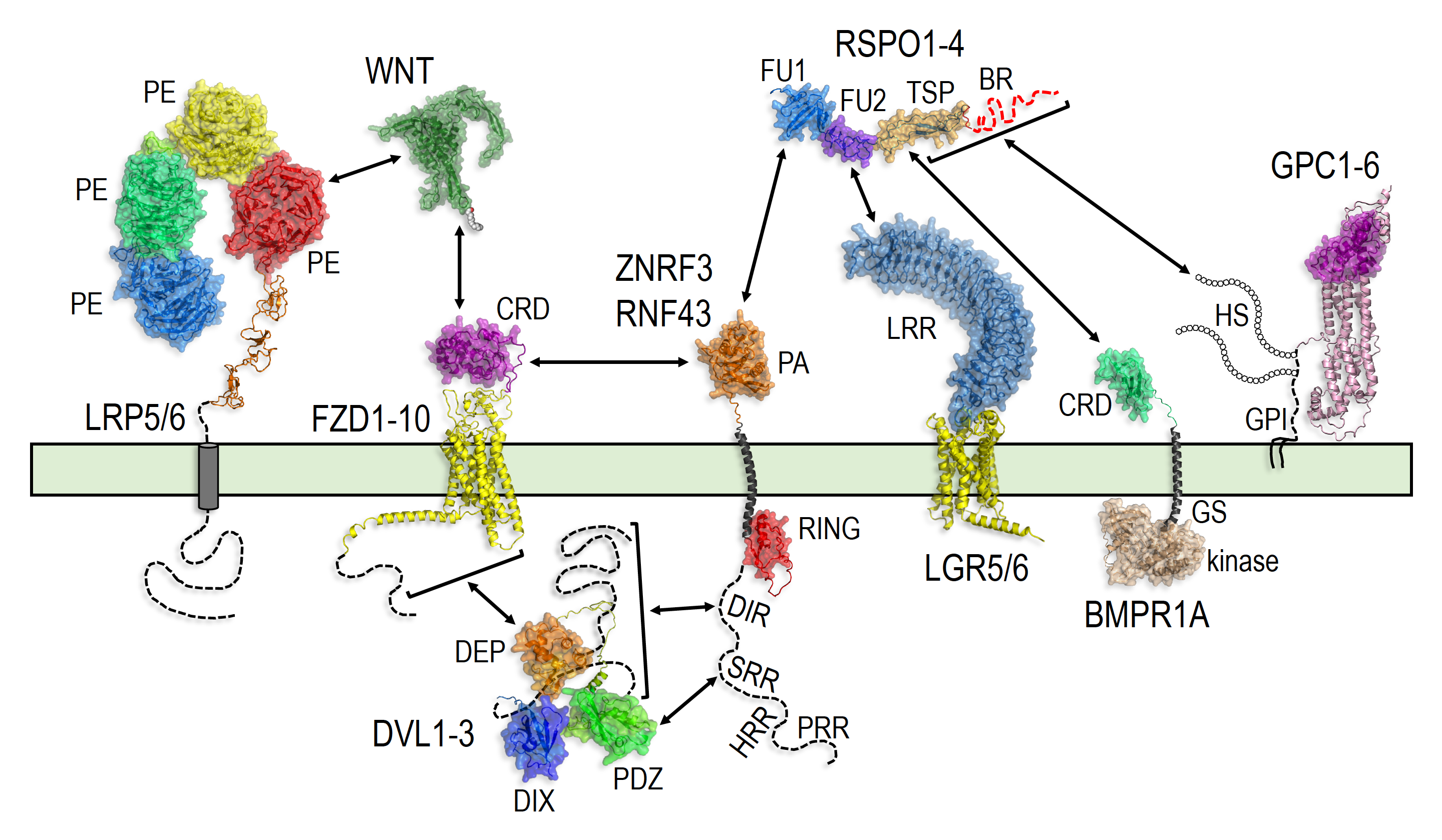
R-spondins are key regulators of WNT signaling strength, but the mechanisms by which they transduce signals are not fully understood and how the four different members of the family control distinct physiological functions is unknown. We discovered that R-spondins 2 and 3 can uniquely potentiate WNT signaling in cells lacking their only known cell-surface receptors, LGRs 4-6, through an alternative interaction with heparan sulfate proteoglycans (HSPGs). LGRs were thought to be required to transduce all R-spondin signals and hence determine their site of action, but now we know that R-spondins can also signal through HSPGs in tissues where LGRs are not expressed. The following research projects are aimed at elucidating the molecular mechanisms and physiological functions of ‘HSPG-dependent’ signaling by R-spondins.
3) Molecular mechanisms of HSPG-dependent signaling by R-spondins.
A central question regarding any signal transduction mechanism is how binding of the ligand to the receptor communicates the signal across the plasma membrane and into the cell. In the prevailing model, simultaneous binding of R-spondins to LGR receptors and to the transmembrane ubiquitin ligases ZNRF3 or RNF43 triggers their internalization. This prevents ZNRF3 and RNF43 from targeting WNT receptors for ubiquitin-mediated degradation, thus increasing their abundance on the cell surface and enhancing sensitivity to WNT ligands. We are investigating the molecular mechanisms whereby R-spondins transduce signals in the absence of LGRs through their alternative HSPG receptors to potentiate WNT signaling.
4) Physiological roles for LGR-dependent and HSPG-dependent signaling by R-spondins during embryonic development and in stem cells.
Gene amplifications followed by neo- or sub-functionalization of different gene family members underlie much of the functional diversification generated during evolution. Yet, paralogues are often deemed to be redundant, and drastically different phenotypes resulting from their loss are simply attributed to tissue-specific patterns of expression. This is the prevailing view regarding the four members of the R-spondin family. However, the markedly distinct phenotypes caused by the loss of different R-spondins or their LGR receptors could also be due to intrinsic differences in the capacity of these R-spondins to signal through LGR-dependent and HSPG-dependent mechanisms. We are investigating in what contexts these two modes of signaling are used during development, tissue homeostasis and regeneration. We are also trying to understand what unique properties make LGR-dependent and HSPG-dependent R-spondin signaling specifically suited to their physiological functions.
Publications
- Bibliography Link
- View Dr. Lebensohn's PubMed Summary.
Receptor control by membrane-tethered ubiquitin ligases in development and tissue homeostasis
Biography

Andres M. Lebensohn, Ph.D.
Andres Lebensohn grew up in Buenos Aires, Argentina. He obtained his B.A. in molecular and cell biology from the University of California at Berkeley, where he conducted undergraduate research in enzymology with Dr. Daniel E. Koshland, Jr. He obtained his Ph.D. in cell and developmental biology from Harvard University, where he used biochemical reconstitution approaches to study regulation of the actin cytoskeleton by the WAVE complex in the laboratory of Dr. Marc W. Kirschner. He then pursued postdoctoral studies as a Helen Hay Whitney Foundation fellow in the laboratory of Dr. Rajat Rohatgi at Stanford University. In close collaboration with Dr. Jan Carette, he conducted reporter-based, forward genetic screens in haploid human cells to uncover new regulatory mechanisms in WNT signaling. Dr. Lebensohn joined the Laboratory of Cellular and Molecular Biology in October of 2018. He lives in Bethesda with his wife Alexandra (who is a cancer genetic counselor at NIH) and their two children, Gabi and Ani (who snuck into some photos in the Lab Life section of the website). When not in the lab, you may find him fishing and crabbing in the Chesapeake Bay, paddleboarding in the Potomac River and biking the Capital Crescent Trail.
Job Vacancies
We have no open positions in our group at this time, please check back later.
To see all available positions at CCR, take a look at our Careers page. You can also subscribe to receive CCR's latest job and training opportunities in your inbox.
Team




News
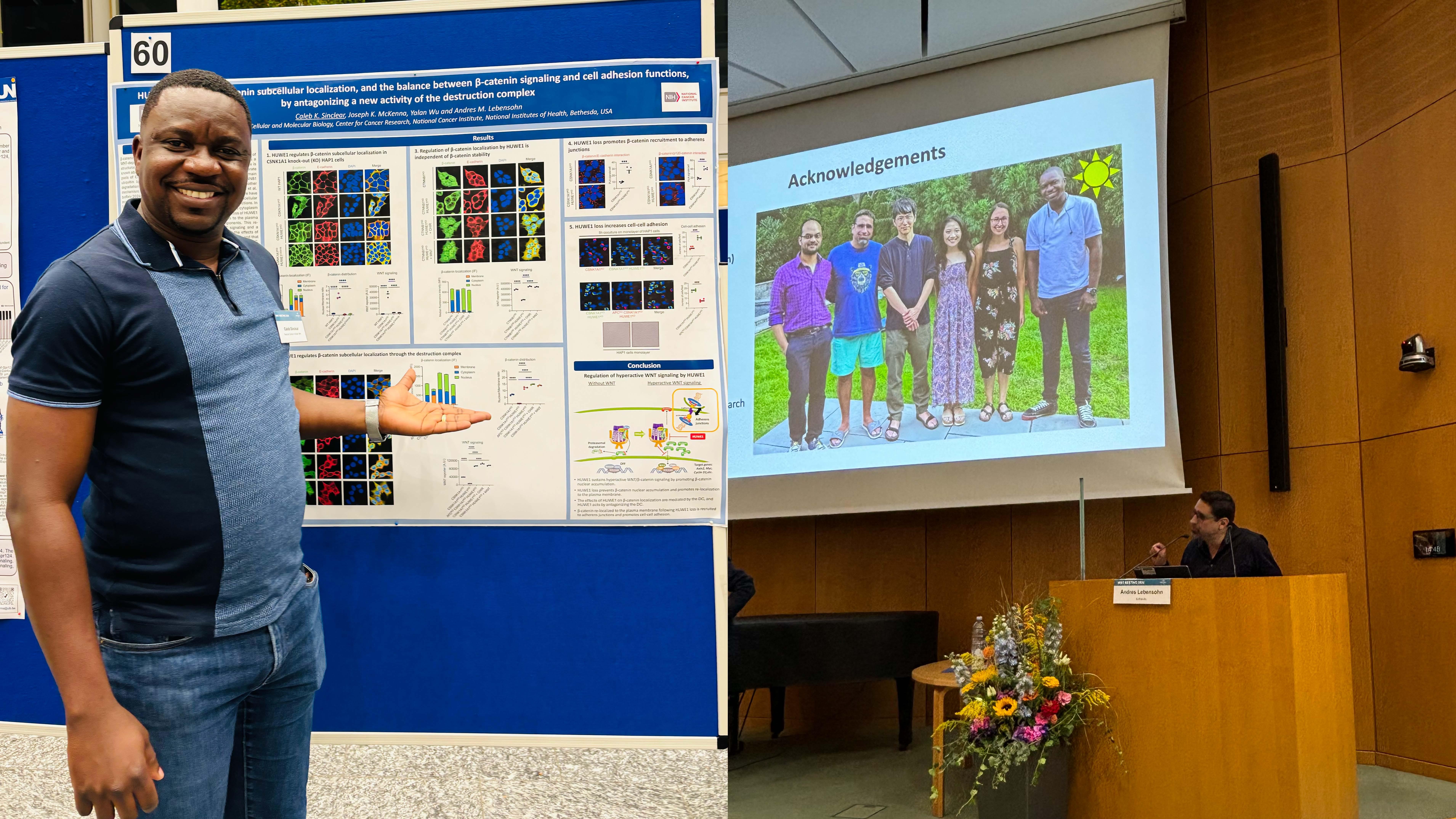
September 25-27, 2024 - Rebecca, Moon, Caleb and Andres attended the WNT Meeting 2024 in Heidelberg, Germany. Caleb presented a poster and Andres gave a talk.
June 3, 2024 - Moon Tran joined the lab as a post-doc. She is originally from Vietnam and obtained her Ph.D. from The Graduate University for Advanced Studies (SOKENDAI) - Japan National Institute for Basic Biology (NIBB) in Japan. Welcome Moon!

April 3, 2024 - As a member and Chair of The Tenure Track Investigator Representatives Committee, Andres received an NCI Director's Award for Emerging Leader-NCI Champions in recognition of "Significant and numerous contributions to improving the experience of Tenure Track Investigators in the NCI Center for Cancer Research."

March 17, 2024 - Our preprint entitled "The ubiquitin ligase HUWE1 enhances WNT signaling by antagonizing destruction complex-mediated β-catenin degradation and through a mechanism independent of β-catenin stability" is on bioRxiv. In this manuscript we demonstrate that the ubiquitin ligase HUWE1 enhances WNT signaling through two distinct mechanisms. First, HUWE1 antagonizes the phosphorylation and degradation of β-catenin by the destruction complex. Second, HUWE1 enhances WNT signaling through a mechanism independent from control of β-catenin stability. The effects of HUWE1 on WNT signaling require its ubiquitin ligase activity and are mediated by a subset of destruction complex components, including APC, AXIN1 and GSK3A/B. Regulation of β-catenin abundance and a downstream step in the pathway by HUWE1, mediated by the destruction complex, would be an efficient way to coordinately control multiple processes that determine WNT signaling output. Congratulations Joey and Yalan, co-first authors, for a beautiful and thorough genetic analysis of the role of HUWE1 in WNT signaling!
September 11, 2023 - Rebecca Kim joined the lab as a post-bac. She comes from Brown University. Welcome Rebecca!

July 30, 2023 - Caleb presented a poster at the Wnt Gordon Research Seminar and Conference in Castelldefels, Spain!
June 28, 2023 - Yalan left the lab to start her M.D./Ph.D. in the Medical Scientist Training Program (MSTP) at University of Maryland School of Medicine in Baltimore. It was a real pleasure having you in the lab (and exploring Japan together!) and we wish you all the best in your future endeavors!
May 22, 2023 - Yalan was recognized as a recipient of the "Outstanding Poster Awards" during NIH Postbac Poster Day 2023. Congratulations Yalan!
March 14, 2023 - Andres presented a seminar at the Swammerdam Institute for Life Sciences, University of Amsterdam in The Netherlands.
March 13, 2023 - Saskia defended her thesis at the University of Amsterdam, including a chapter on the work she did in the lab. Congratulations Saskia!

December 6, 2022 - Andres presented a talk at the Minisimposium on "Mechanisms of intracellular signaling: from receptor to effector" during the Cell Bio 2022 ASCB/EMBO meeting in Washington, D.C.
November 18, 2022 - Yalan presented a poster at the Wnt 2022 EMBO Workshop in Awaji, Japan!

November 7, 2022 - Sara was awarded the competitive NCI Sallie Rosen Kaplan (SRK) Postdoctoral Fellowship for Women Scientists. Congratulations Sara!
September 19, 2022 - Caleb Sinclear joined the lab as a post-doc. He is originally from Ghana and obtained his Ph.D. from Tokyo Medical and Dental University in Japan. Welcome Caleb!

September 7, 2022 - Noa left the lab to gain clinical experience with the group of Dr. Leighton Chan in the Rehabilitation Medicine Department of the NIH Clinical Center. It was great having you in the lab and we wish you all the best in your future endeavors!
August 15, 2022 - Sara Konopelski Snavely joined the lab as a post-doc. She is originally from northern California and obtained her Ph.D. from University of California, Davis, where she did her dissertation in the lab of Dr. Henry Ho. Welcome Sara!

July 26, 2022 - Andres was a judge in the "Genes in Space" contest, which invites students in grades 7 through 12 to design biology experiments that address real-world challenges in space exploration. The five finalists presented their proposals at the International Space Station (ISS) Research and Development Conference in Washington, D.C. Student Pristine Onuoha from Chapel Hill High School in Chapel Hill, NC, was this year's winner. Her experiment seeks to understand the mechanism behind telomere lengthening, a chromosomal change that has been observed in space travelers. Her experiment will be performed by astronauts aboard the International Space Station (ISS) next year.
Andres also delivered the Genes in Space Reception Keynote Address at the Planet Word Museum, in honor of The John Hatch Memorial Prize in Mentorship: "Mentoring: above all, it’s about caring"
June 11, 2022 - Our book chapter entitled "Receptor control by membrane-tethered ubiquitin ligases in development and tissue homeostasis" was published in Volume 150 of Current Topics in Developmental Biology, a volume about "Cell-Cell Signaling in Development" edited by Thomas Kornberg. See the online version here, or download the PDF from the publications section of our website. This chapter explores in great depth an emerging concept in cell signaling: how membrane-tethered ubiquitin ligases control the sensitivity of target cells to secreted ligands by regulating the abundance of signaling receptors at the cell surface. We focus on two examples of this concept: (1) the transmembrane ubiquitin ligases ZNRF3 and RNF43 that regulate WNT and BMP receptor abundance in response to R-spondin ligands and (2) the membrane-recruited ubiquitin ligase MGRN1 that controls Hedgehog and melanocortin receptor abundance. The mechanisms presented in the chapter are illustrated by beautiful structural and protein interaction models enabled by AlphaFold.
September 8, 2021 - Noa Sasson joined the lab as a post-bac. She comes from University of California, Santa Barbara. Welcome Noa!

August 31, 2021 - Saskia finished her Fulbright Scholarship and went back to The Netherlands, where she will start her new Scientist position at OcellO, a biotech company in Leiden. It was awesome having you in the lab! We wish you all the best in your future endeavors.
August 4, 2021 - An interview with Saskia about coming to the NIH as a Visiting Fulbright Scholar - and other thoughts on being a scientist - is featured in Elsevier Connect. Congratulations Saskia!

June 3, 2021 - Joey left us to start his Ph.D. in the Department of Molecular and Cell Biology at UC Berkeley. He will travel cross-country before settling in sunny California. It was fantastic having you in the lab (and fishing monster rock fish together) and we wish you all the best in your future endeavors!
April 5, 2021 - Saskia de Man is visiting the lab as a Fulbright Scholar. She is a Ph.D. student in the lab of Dr. Renée van Amerongen at the University of Amsterdam. Welcome Saskia!

October 13, 2020 - Myungjoo Shin joined the lab as a post-doc. He is originally from Seoul, South Korea and obtained his Ph.D. from Cornell University. Welcome Myungjoo!

June 29, 2020 - Yalan Wu joined the lab as a post-bac. She comes from Boston University. Welcome Yalan!

May 20, 2020 - Our latest paper was published as a Research Advance in eLife! Check it out: https://elifesciences.org/articles/54469. This work builds upon our 2018 eLife Research Article (https://elifesciences.org/articles/33126) to show that interactions between the TSP/BR domain of R-spondin 3 and the heparan sulfate chains of heparan sulfate proteoglycans, such as glypicans and syndecans, are necessary and sufficient to promote LGR-independent potentiation of WNT signaling. Surprisingly, these interactions also contribute substantially to the LGR-dependent signaling by R-spondins necessary to promote the growth of small intestinal organoids. These findings further our understanding of how R-spondins enhance WNT signaling during embryonic development and adult stem cell self-renewal. Any @NCIResearchCtr tweeter followers out there, feel free to retweet :) https://twitter.com/NCIResearchCtr/status/1263904972107726851?s=20
May 14, 2020 - Joey was recognized as a recipient of the "Outstanding Poster Awards" during the first virtual NIH Postbac Poster Day 2020. From the Director of the Office of Intramural Training & Education: "On behalf of the Office of Intramural Training & Education, I want to congratulate you on presenting an outstanding poster at NIH Postbac Poster Day 2020! The judging process was highly competitive, and you should be proud of your accomplishment. This year 839 posters were presented on Poster Day. Of those, 170 were recognized as outstanding, based on the high scores they received during the judging process." Congratulations Joey!
February 3, 2020 - Praveen Sonkusre joined the lab as a post-doc. He comes from CSIR-Institute of Microbial Technology, Chandigarh, India. Welcome Praveen!

January 27, 2020 - Courtney left us to join the lab of Dr. Kathleen Merikangas at the National Institute of Mental Health. She will spend a year doing research on mood disorders before applying to graduate school in public health. It was a pleasure having you as the first post-bac in the lab and we wish you all the best in your future endeavors!
August 19, 2019 - Joey McKenna joined the lab as a post-bac. He comes from UC Berkeley (Andres' Alma Mater). Welcome Joey!

August 10-16, 2019 - Andres and Courtney attended the Gordon Research Conference "Wnt Signaling Networks in Development, Disease and Regeneration" in Mount Snow, West Dover, VT. Andres gave a talk entitled "The Ubiquitin Ligase HUWE1 Regulates Canonical WNT Signaling Independently of Changes in Beta-Catenin Abundance"
July 31, 2019 - Andres was a judge in the "Genes in Space" contest, which invites students in grades 7 through 12 to design biology experiments that address real-world challenges in space exploration. The five finalists presented their proposals at the International Space Station (ISS) Research and Development Conference in Atlanta, GA, and the winning team's experiment will be performed by astronauts aboard the ISS U.S. National Laboratory in 2020.
Check out the Genes in Space website: https://www.genesinspace.org/

The judges with Astronaut Serena Auñón-Chancellor
January 28, 2019 - Courtney Quick joined the lab as the first post-bac. She comes from UNC Asheville. Welcome Courtney!

December 8, 2018 - Andres presented one of the two talks selected as top abstracts at the ASCB Doorstep Meeting "Beyond Homeostasis - Stem Cells Under Stress" in San Diego, CA: “Mechanisms and physiological implications of LGR-dependent and LGR-independent potentiation of WNT signaling by R-spondins”
October 1, 2018 - Andres joined the Laboratory of Cellular and Molecular Biology. He is very excited to begin studies on new regulatory mechanisms of WNT signaling!
Alumni







Lab Life

Rebecca, Moon, Caleb and Andres visiting Heidelberg, Germany during the WNT Meeting 2024. Three generations of the Kirschner lab lineage got together for this momentous occasion, including Marc himself and Henry Ho, Andres' fellow grad student and mentor in the Kirschner lab! Andres got a bit carried away with a classic german dish, pork knuckle... (9/25-27/2024)

Sara, Caleb, Praveen and Andres ventured into the wilderness during a lab outing to Shenandoah National Park. Caleb and Andres even got wasp stings to prove it! (9/16/2023)


Yalan, Sara, Caleb and Andres after completing the NIH Take-a-hike-day walk (6/15/2023)


Ani visiting the lab during take-your-child-to-work day (4/27/2023)


The highlights of our holiday party were decorating cookies and Caleb's words of wisdom (12/19/2022)



The Lebensohn Lab having lunch and bowling at Pike & Rose in North Bethesda, MD (10/13/2022)


The Lebensohn Lab grows! We celebrated Noa's farewell with family and an intense game of cornhole (9/18/2022)

Yalan celebrated her birthday with special guests, Ani and Gabi (7/7/2022)

Lebensohn Lab members stretched their legs by walking 5K during "NIH Take a Hike Day" (6/16/2022)

Joey's farewell celebration with the extended Lebensohn Lab family (6/2/2021)

Praveen, Yalan, Joey and Andres celebrated the two-year anniversary of the Lebensohn Lab with a picnic and fishing in the Chesapeake near Chester River, MD (10/4/2020)

Joey, Gabi, Ani and Courtney hard at work decorating cookies during the Lebensohn Lab Christmas party! (12/19/2019)

Andres, Joey and Courtney celebrating the one-year anniversary of the Lebensohn Lab at Rasika in Washington, D.C. (10/3/2019)

Andres and Courtney at the WNT Gordon Research Conference in Mount Snow, West Dover, VT (8/15/2019)

Courtney's birthday celebration with Gabi and Ani, and vegan cake baked by Alex! (6/26/2019)