
Terrence R. Burke Jr., Ph.D.
- Center for Cancer Research
- National Cancer Institute
- Building 376, Room 210
- Frederick, MD 21702-1201
- 301-846-5906
- burkete@nih.gov
RESEARCH SUMMARY
Dr. Burke utilizes bioorganic and medicinal chemistry to prepare new biologically-active molecules, with an emphasis on peptides and peptide mimetics. His recent work has dealt with the development of inhibitors directed against phosphor-dependent protein-protein interactions, HIV-1 integrase and protein-tyrosine phosphatases. He is also engaged in developing antibody-drug conjugates. Link to additional information about Dr. Burke's research.
Areas of Expertise
Terrence R. Burke Jr., Ph.D.
Research
Bioorganic Medicinal Chemistry and the Modulation of Kinase-Dependent Signal Transduction
Pharmacological agents are being developed to modulate phospho-dependent cell signaling. Historically, our emphasis in this area has been on inhibitors of phosphotyrosyl (pTyr)-dependent binding interactions, which are mediated by src homology 2 (SH2) domains and on protein-tyrosine phosphatase (PTP) inhibitors. More recently, our efforts have focused on developing polo-like kinase 1 (Plk1) polo-box domain (PBD)-binding inhibitors. Central to all these efforts is the development phosphoamino acid surrogates that afford either increased stability toward enzymatic degradation, increased affinity or increased cellular uptake.
In the SH2 domain area, we have collaborated with Dr. Donald Bottaro (CCR, NCI) to develop cell-permeable growth factor receptor-bound 2 (Grb2) antagonists as potential new therapeutics for a variety of erbB-2- and MET-dependent cancers, including breast cancer. For this work, we designed peptidomimetics as conformationally constrained analogs of natural Grb2 SH2 domain-bound pTyr-containing peptides. In related research, a series of new pTyr-mimicking amino acid analogs were prepared to enhance cell permeability.
In the phosphatase area, our recent efforts have concerned the development of inhibitors of the Yersinia pestis phosphatase, YopH, which is a PTP required for infectivity of this potential bioterrorism agent. This work is being conducted as a collaboration with Drs. David Waugh (CCR, NCI) and Robert Ulrich (USAMRIID). Here we have used an oxime library-based approach guided by X-ray co-crystal structures of our inhibitors bound to YopH. In the Plk1 area, we currently have a significant effort underway to develop Plk1 PBD-binding antagonists. This work is being done in collaboration with Dr. Kyung Lee (CCR, NCI). Starting from a pentamer phosphothreonine (pThr) peptide derived from a cognate Plk1 substrate, we are designing and synthesizing peptides and peptide mimetics that contain modified amino acid analogs. We have arrived at agents with extremely high PBD-binding affinity, that have the ability to block division of cancer cells in culture. The goal of this work is to achieve compounds that can exert potent anticancer effects in animal tumor models.
In separate work, inhibitors of HIV integrase are being developed as potential anti-AIDS drugs. Lead inhibitor structures have initially been derived from several sources, including three-dimensional pharmacophore searching of the more than 250,000 compounds contained within the NCI's chemical repository. Promising compounds have been systematically explored through chemical synthesis of analogs to determine structure-activity relationships (SAR) responsible for integrase inhibition. Information generated in this fashion has been applied to the design and preparation of new analogs having higher potency, reduced collateral cytotoxicity, and greater antiviral protective effects in HIV-infected cells.
In collaboration with Drs. Steve Hughes (CCR, NCI) and Yves Pommier (CCR, NCI) we have recently developed non-cytotoxic agents that exhibit anti-viral efficacies in cell culture models of HIV-1 infectivity that equal or exceed the potencies of current clinical IN inhibitors, while retaining greater efficacy against virus harboring major resistant mutant forms of IN. A third area of investigation concerns developing antibody-drug conjugates (ADCs) in collaboration with Dr. Christoph Rader, Scripps, Florida. This work is focused on designing and synthesizing new constructs for attaching drugs and targeting moieties to antibodies.
Publications
- Bibliography Link
- View Dr. Burke's Complete Bibliography at NCBI.
Structural basis for strand-transfer inhibitor binding to HIV intasomes
Identification of a ligand binding hot spot and structural motifs replicating aspects of tyrosyl-DNA phosphodiesterase I (TDP1) phosphoryl recognition by crystallographic fragment cocktail screening
Structure of the Rpn13-Rpn2 complex provides insights for Rpn13 and Uch37 as anticancer targets
A route to imidazolium-containing phosphopeptide macrocycles
HIV-1 integrase strand transfer inhibitors with reduced susceptibility to drug resistant mutant integrases
Biography

Terrence R. Burke Jr., Ph.D.
Dr. Burke received his B.S. in Chemistry from St. Martin's College, followed by his Ph.D. degree from the University of Washington under the direction of Professor Wendel Nelson. He then studied as a Fellow of the Pharmacology Associate Research Training Program in the Laboratory of Dr. Lance Pohl, National Heart Lung and Blood Institute and subsequently under the direction of Dr. Kenner Rice as a Senior Staff Fellow of the National Institute of Diabetes, Digestive and Kidney Diseases. He briefly left the NIH to serve as Principal Chemist of Peptide Technologies Corporation before returning in 1989 as a tenured Principal Investigator in the Chemical Biology Laboratory (previously, the Laboratory of Medicinal Chemistry). In 2002 he became Head of the Bioorganic and Medicinal Chemistry Section and in 2003 he was appointed a member of the Senior Biomedical Research Service (SBRS).
Job Vacancies
We have no open positions in our group at this time, please check back later.
To see all available positions at CCR, take a look at our Careers page. You can also subscribe to receive CCR's latest job and training opportunities in your inbox.
Team






Covers

Application of a bivalent “click” approach to target tyrosyl-DNA phosphodiesterase 1 (TDP1)
Although inhibiting the DNA repair enzyme tyrosyl-DNA phosphodiesterase 1 (TDP1) synergizes with topoisomerase type I (TOP1) inhibitors in anticancer therapy, development of TDP1 inhibitors has been highly challenging. This may be due to the open and shallow nature of the TDP1 catalytic site and the necessity of competing with a large and highly extended substrate. The toolbox available to chemical biologists for studying TDP1 could be significantly enhanced by introducing the ability to selectively eliminate TDP1 using protein degraders. Our current work starts from phenyl imidazopyridine-based TDP1 inhibitors previously developed from small molecule microarrays (SMMs). Using crystal structures of lead inhibitors bound to TDP1, we designed and synthesized a series of bivalent proteolysis-targeting chimeras (PROTACs). The focus of our current work is to explore synthetic approaches that permit installation of E3 ligase-targeting functionality, while retaining the TDP1 binding. We employed copper-catalyzed azide-alkyne cycloaddition (CuAAC) “click” reactions to assemble PROTAC constituents with 1,2,3-triazolecontaining linkers. With the addition of the relatively large parts of the linkers and E3-targeting moieties, we retained the ability to inhibit TDP1. The successful development of TDP1-directed PROTACS would yield a new therapeutic class that could potentially enhance the efficacy and selectivity of TOP1 inhibitors including those used as payloads in antibody drug conjugates (ADCs).
Xue Zhi Zhao, Wenjie Wang, Md Rasel Al Mahmud, Keli Agama, Yves Pommier and Terrence R. Burke, Jr.
RSC Medicinal Chemistry, 2025, 16, 1969-1985.

Affinity enhancement of polo-like kinase 1 polo box domain-binding ligands by a bivalent approach using a covalent kinase-binding component
The polo-like kinase 1 (Plk1) is an important cell cycle regulator that is recognized as a target molecule
for development of anti-cancer agents. Plk1 consists of a catalytic kinase domain (KD) and a polo-box
domain (PBD), which engages in protein–protein interactions (PPIs) essential to proper Plk1 function.
Recently, we developed extremely high-affinity PBD-binding inhibitors based on a bivalent approach
using the Plk1 KD-binding inhibitor, BI2536, and a PBD-binding peptide. Certain of the resulting bivalent
constructs exhibited more than 100-fold Plk1 affinity enhancement relative to the best monovalent
PBD-binding ligands. Herein, we report an extensive investigation of bivalent ligands that utilize the nonselective
kinase inhibitor Wortmannin as a Plk1 KD-binding component. We found that bivalent ligands
incorporating Wortmannin demonstrated affinity enhancements that could be similar to what we had
obtained with BI2536 and that they could tightly bind to the protein. This suggests that these tight
binding ligands might be useful for structural analysis of full-length Plk1.
Kohei Tsuji, Hirokazu Tamamura and Terrence R. Burke Jr.
RSC Chemical Biology, 2024, 5, 721-728

Identification of multidentate tyrosyl-DNA phosphodiesterase 1 (TDP1) inhibitors that simultaneously access the DNA, protein and catalytic-binding sites by oxime diversification
Tyrosyl-DNA phosphodiesterase 1 (TDP1) is a member of the phospholipase D family that can downregulate the anticancer effects of the type I topoisomerase (TOP1) inhibitors by hydrolyzing the 30-phosphodiester bond between DNA and the TOP1 residue Y723 in the critical stalled intermediate that is the foundation of TOP1 inhibitor mechanism of action. Thus, TDP1 antagonists are attractive as potential enhancers of TOP1 inhibitors. However, the open and extended nature of the TOP1–DNA substratebinding region has made the development of TDP1 inhibitors extremely challenging. In this study, starting from our recently identified small molecule microarray (SMM)-derived TDP1-inhibitory imidazopyridine motif, we employed a click-based oxime protocol to extend the parent platform into the DNA and TOP1 peptide substrate-binding channels. We applied one-pot Groebke Blackburn–Bienayme multicomponent reactions (GBBRs) to prepare the needed aminooxy-containing substrates. By reacting these precursors with approximately 250 aldehydes in microtiter format, we screened a library of nearly 500 oximes for their TDP1 inhibitory potencies using an in vitro florescence-based catalytic assay. Select hits were structurally explored as their triazole- and ether-based isosteres. We obtained crystal structures of two of the resulting inhibitors bound to the TDP1 catalytic domain. The structures reveal that the inhibitors form hydrogen bonds with the catalytic His-Lys-Asn triads (‘‘HKN’’ motifs: H263, K265, N283 and H493, K495, N516), while simultaneously extending into both the substrate DNA and TOP1 peptide-binding grooves. This work provides a structural model for developing multivalent TDP1 inhibitors capable of binding in a tridentate fashion with a central component situated within the catalytic pocket and extensions that project into both the DNA and TOP1 peptide substrate-binding regions.
Xue Zhi Zhao, Wenjie Wang, George T. Lountos, Evgeny Kiselev, Joseph E. Trope, Danielle Needle, Yves Pommier and Terrence R. Burke, Jr.
RSC Chemical Biology, 2023, 4, 334-343

Synthetic Approaches to a Key Pyridone-carboxylic Acid Precursor Common to the HIV‑1 Integrase Strand Transfer Inhibitors Dolutegravir, Bictegravir, and Cabotegravir
Dolutegravir (DTG), Bictegravir (BIC), and Cabotegravir (CAB) are the second-generation integrase strand transfer inhibitors (INSTIs) that have been FDA-approved for the treatment of HIV-1 infection. Preparation of these INSTIs utilizes the common intermediate 1-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-1,4-dihydropyridine-3-carboxylic acid (6). Presented herein is a literature and patent review of synthetic routes used to access the pharmaceutically important intermediate 6. The review highlights the ways in which small fine-tuned synthetic modifications have been used to achieve good yields and regioselectivity of ester hydrolysis.
Pankaj S. Mahajan and Terrence R. Burke, Jr.
Org. Process Res. Dev. 2023, 27, 5, 847–853

Development of ultra-high affinity bivalent ligands targeting the polo-like kinase 1
Back Cover
Showcasing research from the Burke Group in the Chemical Biology Laboratory, NCI, NIH, USA. Development of ultra-high affinity bivalent ligands targeting the polo-like kinase 1 Bivalent ligands are described that are designed to access both the catalytic kinase domain (KD) and the polo-box domain (PBD) of the polo-like kinase 1 (Plk1). Exceptionally high binding affinities were retained even with minimal linkers between KD and PBD-binding components. This potentially suggests spatial orientation of the KD and PBD consistent with simultaneous occupancy. Such an orientation could have important implications for Plk1 structure-function.
Kohei Tsuji, David Hymel, Buyong Ma, Hirokazu Tamamura, Ruth Nussinov and Terrence R. Burke Jr.
RSC Chemical Biology, 2022, 3 , 1111-1120.

Design and synthesis of a new orthogonally protected glutamic acid analog and its use in the preparation of high affinity polo-like kinase 1 polo-box domain – binding peptide macrocycles
Targeting protein – protein interactions (PPIs) has emerged as an important area of discovery for anticancer therapeutic development. In the case of phospho-dependent PPIs, such as the polo-like kinase 1(Plk1) polo-box domain (PBD), a phosphorylated protein residue can provide high-affinity recognition and binding to target protein hot spots. Developing antagonists of the Plk1 PBD can be particularly challenging if one relies solely on interactions within and proximal to the phospho-binding pocket. Fortunately, the affinity of phosphor dependent PPI antagonists can be significantly enhanced by taking advantage of interactions in both the phospho-binding site and hidden “cryptic” pockets that may be revealed on ligand binding. In our current paper, we describe the design and synthesis of macrocyclic peptide mimetics directed against the Plk1 PBD, which are characterized by a new glutamic acid analog that simultaneously serves as a ring-closing junction that provides accesses to a cryptic binding pocket, while at the same time achieving proper orientation of a phosphothreonine (pT) residue for optimal interaction in the signature phospho-binding pocket. Macrocycles prepared with this new amino acid analog introduce additional hydrogen-bonding interactions not found in the open-chain linear parent peptide. It is noteworthy that this new glutamic acid-based amino acid analog represents the first example of extremely high affinity ligands where access to the cryptic pocket from the pT-2 position is made possible with a residue that is not based on histidine. The concepts employed in the design and synthesis of these new macrocyclic peptide mimetics should be useful for further studies directed against the Plk1 PBD and potentially for ligands directed against other PPI targets.
This article can be accessed directly from the Royal Society of Chemistry at Design and synthesis of a new orthogonally protected glutamic acid analog and its use in the preparation of high affinity polo-like kinase 1 polo-box domain – binding peptide macrocycles - Organic & Biomolecular Chemistry (RSC Publishing)
David Hymel, Kohei Tsuji, Robert A. Grant, Ramesh M. Chingle, Dominique L. Kunciw, Michael B. Yaffe and Terrence R. Burke, Jr.*
Organic & Biomolecular Chemistry, 2021, 19, 7843-7854.

HIV‑1 Integrase Inhibitors with Modifications That Affect Their Potencies against Drug Resistant Integrase Mutants
The inside cover shows INSTIs bound to HIV-1 IN. Compound 5j (magenta) was docked onto the structures of 4d (yellow) and BIC (green) bound to the active site of HIV-1 IN; the surface of IN is shown in white. The surface envelope of the unprocessed 3′ end of the vDNA is shown as brown mesh. Ordered water molecules bound to the active site of IN in the absence of a bound INSTI are shown in cyan.
Abstract: Integrase strand transfer inhibitors (INSTIs) block the integration step of the retroviral lifecycle and are first-line drugs used for the treatment of HIV-1/AIDS. INSTIs have a polycyclic core with heteroatom triads, chelate the metal ions at the active site, and have a halobenzyl group that interacts with viral DNA attached to the core by a flexible linker. The most broadly effective INSTIs inhibit both wild-type (WT) integrase (IN) and a variety of well-known mutants. However, because there are mutations that reduce the potency of all the available INSTIs, new and better compounds are needed. Models based on recent structures of HIV-1 and red-capped mangabey SIV INs suggest modifications in the INSTI structures that could enhance interactions with the 3′-terminal adenosine of the viral DNA, which could improve performance against INSTI resistant mutants. We designed and tested a series of INSTIs having modifications to their naphthyridine scaffold. One of the new compounds retained good potency against an expanded panel of HIV-1 IN mutants that we tested. Our results suggest the possibility of designing inhibitors that combine the best features of the existing compounds, which could provide additional efficacy against known HIV-1 IN mutants.
Steven J. Smith, Xue Zhi Zhao, Dario Oliveira Passos, Valerie E. Pye, Peter Cherepanov, Dmitry Lyumkis, Terrence R. Burke, Jr., and Stephen H. Hughes*
ACS Infectious Diseases, 2021, 7, 1469-1482

Small molecule microarray identifies inhibitors of tyrosyl-DNA phosphodiesterase 1 that simultaneously access the catalytic pocket and two substrate binding sites
We now report examining a 21 000-member library of drug-like Small Molecules in Microarray (SMM) format for their ability to bind Alexa Fluor 647 (AF647)-labeled TDP1. The screen identified structurally similar N,2-diphenylimidazo[1,2-a]pyrazin-3-amines as TDP1 binders and catalytic inhibitors. We then explored the core heterocycle skeleton using one-pot Groebke–Blackburn–Bienayme multicomponent reactions and arrived at analogs having higher inhibitory potencies. Solving TDP1 co-crystal structures of a subset of compounds showed their binding at the TDP1 catalytic site, while mimicking substrate interactions. Although our original fragment screen differed significantly from the current microarray protocol, both methods identified ligand–protein interactions containing highly similar elements. Importantly inhibitors identified through the SMM approach show competitive inhibition against TDP1 and access the catalytic phosphate-binding pocket, while simultaneously providing extensions into both the substrate DNA and peptide-binding channels. As such, they represent a platform for further elaboration of trivalent ligands, that could serve as a new genre of potent TDP1 inhibitors
Xue Zhi Zhao, Evgeny Kiselev, George T. Lountos, Wenjie Wang, Joseph E. Tropea, Danielle Needle, Thomas A. Hilimire, John S. Schneekloth, Jr, David S. Waugh, Yves Pommier and Terrence R. Burke, Jr.
Chemical Science, 2021, 12, 3876 - 3884

A new genre of fluorescence recovery assay to evaluate polo-like kinase 1 ATP-competitive inhibitors
Using a probe consisting of a fluorescein-labeled variant of the potent polo-like kinase 1 (Plk1) inhibitor BI2536 [FITC-PEG-Lys(BI2536) 4], we were able to determine half maximal inhibitory concentration (IC50) of ATP-competitive Type 1 inhibitors of Plk1 by means of a fluorescence recovery assay. This methodology represents a cost-effective and simple alternative to traditional kinase assays for initial screening of potential Plk1 inhibitors.
This article can be accessed directly from the Royal Society of Chemistry at https://pubs.rsc.org/en/content/articlelanding/2020/AY/D0AY01223H#!divAbstract
Kohei Tsuji, David Hymel and Terrence R. Burke, Jr.
Analytical Methods, 2020, 12, 4418-4421.

Application of Post Solid-Phase Oxime Ligation to Fine-Tune Peptide–Protein Interactions
Protein–protein interactions (PPIs) represent an extremely attractive class of potential new targets for therapeutic intervention; however, the shallow extended character of many PPIs can render developing inhibitors against them as exceptionally difficult. Yet this problem can be made tractable by taking advantage of the fact that large interacting surfaces are often characterized by confined “hot spot” regions, where interactions contribute disproportionately to overall binding energies. Peptides afford valuable starting points for developing PPI inhibitors because of their high degrees of functional diversity and conformational adaptability. Unfortunately, contacts afforded by the 20 natural amino acids may be suboptimal and inefficient for accessing both canonical binding interactions and transient “cryptic” binding pockets. Oxime ligation represents a class of biocompatible “click” chemistry that allows the structural diversity of libraries of aldehydes to be rapidly evaluated within the context of a parent oxime-containing peptide platform. Importantly, oxime ligation represents a form of post solid-phase diversification, which provides a facile and empirical means of identifying unanticipated protein–peptide interactions that may substantially increase binding affinities and selectivity. The current review will focus on the authors’ use of peptide ligation to optimize PPI antagonists directed against several targets, including tumor susceptibility gene 101 (Tsg101), protein tyrosine phosphatases (PTPases) and the polo-like kinase 1 (Plk1). This should provide insights that can be broadly directed against an almost unlimited range of physiologically important PPIs.
Xue Zhi Zhao, Fa Liu and Terrence R. Burke Jr.
Molecules, 2020, 25 (12), 2807.

Identification of a ligand binding hot spot and structural motifs replicating aspects of tyrosyl-DNA phosphodiesterase I (TDP1) phosphoryl recognition by crystallographic fragment cocktail screening
Tyrosyl DNA-phosphodiesterase I (TDP1) repairs type IB topoisomerase (TOP1) cleavage complexes generated by TOP1 inhibitors commonly used as anticancer agents. TDP1 also removes DNA 3′ end blocking lesions generated by chain-terminating nucleosides and alkylating agents, and base oxidation both in the nuclear and mitochondrial genomes. Combination therapy with TDP1 inhibitors is proposed to synergize with topoisomerase targeting drugs to enhance selectivity against cancer cells exhibiting deficiencies in parallel DNA repair pathways. A crystallographic fragment screening campaign against the catalytic domain of TDP1 was conducted to identify new lead compounds. Crystal structures revealed two fragments that bind to the TDP1 active site and exhibit inhibitory activity against TDP1. These fragments occupy a similar position in the TDP1 active site as seen in prior crystal structures of TDP1 with bound vanadate, a transition state mimic. Using structural insights into fragment binding, several fragment derivatives have been prepared and evaluated in biochemical assays. These results demonstrate that fragment-based methods can be a highly feasible approach toward the discovery of small-molecule chemical scaffolds to target TDP1, and for the first time, we provide co-crystal structures of small molecule inhibitors bound to TDP1, which could serve for the rational development of medicinal TDP1 inhibitors.
George T. Lountos, Xue Zhi Zhao, Evgeny Kiselev, Joseph E. Tropea, Danielle Needle, Yves Pommier, Terrence R. Burke, Jr. and David S. Waugh
Nucleic Acids Research, 2019, 47, 10134–10150.

Development of Highly Selective 1,2,3-Triazole-containing Peptidic Polo-like Kinase 1 Polo-box Domain-binding Inhibitors
Cover Story: Two highly selective triazole-containing peptidic polo-like kinase 1 (Plk1) polo-box domain (PBD)-binding inhibitors (3d in green and 4b in orange) are depicted, binding within the hydrophobic “cryptic” binding pocket on the surface of the Plk1 PBD (light blue). The placement was guided by our previously reported X-ray crystal structure of PBD-bound parent peptide (2a) (PDB accession code: 3RQ7), whose binding pocket is highlighted in yellow. These ligands retain the high PBD-binding affinity of the parent peptide, while showing desirable enhanced selectivity for the PBD of Plk1 relative to the PBDs of Plk2 and Plk3.
Members of the polo-like kinase (Plk) family of serine/threonine protein kinases play crucial roles in cell cycle regulation and proliferation. Of five Plks (Plk1 – 5), Plk1 is recognized as an anticancer drug target. Plk1 contains multiple structural components that are important for its proper biological function. These include an N-terminal catalytic domain and a C-terminal non-catalytic polo-box domain (PBD). The PBD binds to phosphothreonine (pT) and phosphoserine-containing sequences. Blocking PBD-dependent interactions offers a potential means of down-regulating Plk1 function that is distinct from targeting its ATP-binding site. Previously, we demonstrated by tethering alkylphenyl chains from the N(π)-position of the His residue in the 5-mer PLHSpT, that we were able to access a hydrophobic "cryptic" binding pocket on the surface of the PBD, and in so doing enhance binding affinities by approximately 1000-fold. More recently, we optimized these PBD-ligand interactions using an oxime ligation-based strategy. Herein, using azide-alkyne cycloaddition reactions, we explore new triazole-containing PBD-binding antagonists. Some of these ligands retain the high PBD-binding affinity of the parent peptide, while showing desirable enhanced selectivity for the PBD of Plk1 relative to the PBDs of Plk2 and Plk3.
Xue Zhi Zhao, Kohei Tsuji, David Hymel and Terrence R. Burke Jr.
Molecules 2019, 24, 1488

Histidine N(τ)-cyclized macrocycles as a new genre of polo-like kinase 1 polo-box domain-binding inhibitors
Transition toward peptide mimetics of reduced size is an important objective of peptide macrocyclization. We have previously shown that PLH∗SpT (2a) (where H∗ indicates the presence of a –(CH2)8Ph group at the N(π) position and pT indicates phosphothreonine) is an extremely high affinity ligand of the polo-like kinase 1 (Plk1) polo-box domain (PBD). Herein we report that C-terminal macrocyclization of 2a employing N(π),N(τ)-bis-alkylated His residues as ring junctions can be achieved in a very direct fashion. The resulting macrocycles are highly potent in biochemical assays and maintain good target selectivity for the Plk1 PBD versus the PBDs of Plk2 and Plk3. Importantly, as exemplified by 5d, our current approach permits deletion of the N-terminal “Pro-Leu” motif to yield tripeptide ligands with decreased molecular weight, which retain high affinity and show improved target selectivity. These findings could fundamentally impact the future development of peptide macrocycles in general and Plk1 PBD-binding peptide mimetics in particular.
David Hymel, Robert A. Grant, Kohei Tsuji, Michael B. Yaffe and Terrence R. Burke
Bioorganic & Medicinal Chemistry Letter, 2018, 28 (19), 3202-3205.

HIV-1 Integrase-Targeted Short Peptides Derived from a Viral Protein R Sequence
HIV-1 integrase (IN) inhibitors represent a new class of highly effective anti-AIDS therapeutics. Current FDA-approved IN strand transfer inhibitors (INSTIs) share a common mechanism of action that involves chelation of catalytic divalent metal ions. However, the emergence of IN mutants having reduced sensitivity to these inhibitors underlies efforts to derive agents that antagonize IN function by alternate mechanisms. Integrase along with the 96-residue multifunctional accessory protein, viral protein R (Vpr), are both components of the HIV-1 pre-integration complex (PIC). Coordinated interactions within the PIC are important for viral replication. Herein, we report a 7-mer peptide based on the shortened Vpr (69–75) sequence containing a biotin group and a photo-reactive benzoylphenylalanyl residue, and which exhibits low micromolar IN inhibitory potency. Photo-crosslinking experiments have indicated that the peptide directly binds IN. The peptide does not interfere with IN-DNA interactions or induce higher-order, aberrant IN multimerization, suggesting a mode of action for the peptide that is distinct from clinically used INSTIs and developmental allosteric IN inhibitors. This compact Vpr-derived peptide may serve as a valuable pharmacological tool to identify a potential new pharmacologic site.
Xue Zhi Zhao, Mathieu Métifiot, Evgeny Kiselev, Jacques J. Kessi, Kasthuraiah Maddali, Christophe Marchand, Mamuka Kvaratskhelia, Yves Pommier and Terrence R. Burke Jr.
Molecules, 2018, 23 (8), 1858.

Structure-Guided Optimization of HIV Integrase Strand Transfer Inhibitors
Integrase mutations can reduce the effectiveness of the first-generation FDA-approved integrase strand transfer inhibitors (INSTIs), raltegravir (RAL) and elvitegravir (EVG). The second-generation agent, dolutegravir (DTG), has enjoyed considerable clinical success; however, resistancecausing mutations that diminish the efficacy of DTG have appeared. Our current findings support and extend the substrate envelope concept that broadly effective INSTIs can be designed by filling the envelope defined by the DNA substrates. Previously, we explored 1-hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxamides as an INSTI scaffold, making a limited set of derivatives, and concluded that broadly effective INSTIs can be developed using this scaffold. Herein, we report an extended investigation of 6-substituents as well the first examples of 7-substituted analogues of this scaffold. While 7-substituents are not well-tolerated, we have identified novel substituents at the 6-position that are highly effective, with the best compound (6p) retaining better efficacy against a broad panel of known INSTI resistant mutants than any analogues we have previously described. View the article.
Xue Zhi Zhao, Steven J. Smith, Daniel P. Maskell, Mathieu Metífiot, Valerie E. Pye, Katherine Fesen, Christophe Marchand, Yves Pommier, Peter Cherepanov, Stephen H. Hughes, and Terrence R. Burke, Jr.
J. Med. Chem. 2017, 60, 7315-7332.

Application of oxime-diversification to optimize ligand interactions within a cryptic pocket of the polo-like kinase 1 polo-box domain
By a process involving initial screening of a set of 87 aldehydes using an oxime ligation-based strategy, we were able to achieve a several-fold affinity enhancement over one of the most potent previously known polo-like kinase 1 (Plk1) polo-box domain (PBD) binding inhibitors. This improved binding may result by accessing a newly identified auxiliary region proximal to a key hydrophobic cryptic pocket on the surface of the protein. Our findings could have general applicability to the design of PBD-binding antagonists.
Cover design by Joseph Myer
Xue Zhi Zhao, David Hymel and Terrence R. Burke, Jr.
Bioorganic & Medicinal Chemistry Letters, 2016, 26, 5009-5012.

HIV‑1 Integrase Strand Transfer Inhibitors with Reduced Susceptibility to Drug Resistant Mutant Integrases
On the cover: Mutant forms of HIV-1 IN reduce the therapeutic effectiveness of integrase strand transfer inhibitors (INSTIs). The cover figure shows the IN of prototype foamy virus complexed to a novel INSTI (gold) that retains potency against resistant mutants of HIV-1 IN. Overlain are the host and viral DNA substrates (blue and green, respectively), showing substrate mimicry by the inhibitor.
Cover design by Joseph Myer
Xue Zhi Zhao, Steven J. Smith, Daniel P. Maskell, Mathieu Metifiot, Valerie E. Pye, Katherine Fesen, Christophe Marchand, Yves Pommier, Peter Cherepanov, Stephen H. Hughes and Terrence R. Burke, Jr.
ACS Chemical Biology, 2016, 11 , 1074-1081.

Neighbor-Directed Histidine N (s)–Alkylation: A Route to Imidazolium-Containing Phosphopeptide Macrocycles-Biopolymers
Our recently discovered, selective, on-resin route to N(s)-alkylated imidazolium-containing histidine residues affords new strategies for peptide mimetic design. In this, we demonstrate the use of this chemistry to prepare a series of macrocyclic phosphopeptides, in which imidazolium groups serve as ring-forming junctions. Interestingly, these cationic moieties subsequently serve to charge-mask the phosphoamino acid group that directed their formation. Neighbor-directed histidine N(s)-alkylation opens the door to new families of phosphopeptidomimetics for use in a range of chemical biology contexts.
See also: Editorial Information
Wen-Jian Qian, Jung-Eun Park, Robert Grant, Christopher C. Lai, James A. Kelley, Michael B. Yaffe, Kyung S. Lee and Terrence R. Burke Jr.
Peptide Science, 2015, 104, 663–673.

Peptoid–Peptide Hybrid Ligands Targeting the Polo Box Domain of Polo-Like Kinase 1k
The cover picture shows the binding of a PLHSpT derivative, 6q, to the polo-like kinase 1 (Plk1) polo-box domain (PBD), thereby uncovering a new hydrophobic channel (magnified upper right), which is absent in the unliganded protein (magnified lower left). The authors explain how, as a consequence of the additional interaction with the channel, the peptide binds to the Plk1 PBD with a binding affinity more than two orders of magnitude higher. The background image of a stained cell nucleus depicts how this binding results in interference with Plk1 (red dots)-dependent bipolar spindle formation (green), and this ultimately leads to mitotic block and apoptotic cell death in cultured cancer cells.
Fa Liu, Jung-Eun Park, Wen-Jian Qian, Dan Lim, Andrej Scharow, Thorsten Berg, Michael B. Yaffe, Kyung S. Lee*, and Terrence R. Burke Jr.
ChemBioChem , 2012, 13, 1291-1296.

Application of Ring-Closing Metathesis to Grb2 SH3 Domain-Binding Peptides
In silico-generated hypothetical interactions of a ring-closing metathesis-macrocylized peptide bound to the amino terminal SH3 domain of the growth factor receptor bound protein 2 (Grb2). The complex was derived from the NMR solution structure of the bound parent peptide, Ac-V-P-P-P-V-P-P-R-R-R-amide (Protein Data Bank: 3GBQ). The protein surface is shown as electrostatic potential (blue = positive; red = negative).
Fa Liu, Alessio Giubellino, Philip C. Simister, Wenjian Qian, Michael C. Giano, Stephan M. Feller, Donald P. Bottaro and Terrence R. Burke Jr.
Peptide Science, 2011, 96 (6), 780–788.

Discovery of a small-molecule HIV-1 integrase inhibitor-binding site
The lowest energy-binding conformation of an inhibitor bound to the dimeric interface of HIV-1 integrase core domain. The yellow region represents a unique allosteric binding site identified by affinity labeling and mass spectrometry and validated through mutagenesis. This site can provide a potential platform for the rational design of inhibitors selective for disruption of integrase multimerization.
Laith Q. Al-Mawsawi, Valery Fikkert, Raveendra Dayam, Myriam Witvrouw, Terrence R. Burke, Jr., Christoph H. Borchers and Nouri Neamati
PNAS 2006, 103 (26), 10080-10085.
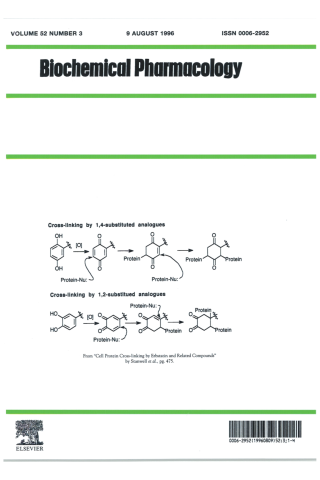
Cell protein cross-linking by erbstatin and related compounds
The scheme depicts a possible mechanism of cross-linking by erbstatin and related analogues. A mechanism of action is proposed which involves initial oxidation to reactive quinone intermediates that subsequently cross-link protein nucleophiles via multiple 1,4-Michael-type additions. Similar alkylation of protein by protein-tyrosine kinase inhibitors, such as herbimycin A, has been invoked.
Caroline Stanwell, Bin Ye, Stuart H Yuspa and Terrence R Burke Jr.
Biochemical Pharmacology 1996, 52 (3), 475-480.
Events
Link to Dr. Burke's 2019 Hillebrand Award Address (December 9, 2020)https://capitalchemist.org/2021/01/join-us-december-9-2020-at-1200-noon-for-presentation-of-the-hillebrand-prize-and-gordon-award/
News
Learn more about CCR research advances, new discoveries and more
on our news section.