
George T. Lountos, Ph.D.
- Center for Cancer Research
- National Cancer Institute
- Bldg. 538, Room 218
- Frederick, MD 21702-1201
- 301-846-5435
- lountosg@mail.nih.gov
RESEARCH SUMMARY
Dr. Lountos is currently involved in a number of highly collaborative research projects within NCI at Frederick involving the structural determination of proteins engaged in cancer and infectious diseases with particular emphasis on the structure-based design of novel small molecule inhibitors. A variety of targets are currently under investigation, including protein kinases and phosphatases, viral proteases, the SUMO-conjugating enzyme, Ubc9, tyrosyl-DNA phosphodiesterase I (TDP1) and other enzymes. Dr. Lountos determined and published the first crystal structure of the Middle East respiratory syndrome coronavirus 3CL protease in 2015 (http://www.ncbi.nlm.nih.gov/pubmed/25945576). Dr. Lountos played a pivotal role in determining high-resolution crystal structures of human checkpoint kinase 2 (Chk2) in complex with various inhibitors that have informed the rational design of potent and specific inhibitors with anti-cancer activity (https://pubmed.ncbi.nlm.nih.gov/19741151/). In collaboration with several NCI laboratories, Dr. Lountos has successfully applied fragment-based drug discovery to identify and structurally characterize the first small molecule scaffolds bound to the cancer drug targets, Ubc9 (https://pubmed.ncbi.nlm.nih.gov/27038327/) and TDP1 (https://pubmed.ncbi.nlm.nih.gov/31199869/) by high resolution X-ray crystallography. Current research is also focused on structural studies of the E. coli LonA protease using single particle cryo-electron microscopy. Our research has been highlighted on the covers of the journals Nucleic Acids Research, Royal Society of Chemistry Chemical Science, Royal Society of Chemistry Chemical Biology, Chemical Biology & Drug Design, and ACS Biochemistry.
Areas of Expertise
Research
Structure-Based Drug Design:
A variety of drug targets involved in cancer and infectious disease are currently being studied by X-ray crystallography in collaboration with medicinal chemists and biochemists within the CCR. Our goal is to elucidate the structural basis for inhibitor binding as a first step towards the optimization of these inhibitors by rational design. In addition to studying protein-inhibitor complexes of compounds identified from high-throughput screening, our group is leading fragment-based drug discovery efforts for drug targets by screening libraries of small drug-like fragments by crystallographic methods. X-ray crystallography is a very powerful tool that can be used to identify and confirm binding of low-affinity fragments to proteins that might not be easliy detected by other screening methods. Structural information gleaned from these studies can be used to optimize low-affinity fragments into inhibitors with higher potency while incorporating favorable drug-like properties.
Current targets under collaborative investigation using structure-based drug design methods include the SUMO-conjugating enzyme, Ubc9 (Dr. Jay Schneekloth, Chemical Biology Laboratory) and tyrosyl-DNA phosphodiesterase I (TDP1) (Dr. Yves Pommier, LMP, Dr. Terrence Burke, CBL). We have also determined the first high-resolution crystal structure of the main protease (3CLpro) from the highly pathogenic Middle East Respiratory Syndrome (MERS) coronavirus in collaboration with Dr. Robert Ulirch, USAMRIID. In collaboration with Dr. Terrence Burke (Chemical Biology Laboratory) and Dr. Robert Ulrich (USAMRIID), we have used structure-based design methods to develop highly potent and specific inhibitors of the Yersinia pestis protein tyrosine phosphatase, YopH (https://pubmed.ncbi.nlm.nih.gov/21443195/). I have been involved in the structure-based design of human checkpoint kinase 2 inhibitors in collaboration with Dr. Yves Pommier and Dr. Robert Shoemaker (NCI Division of Cancer Prevention) (https://pubmed.ncbi.nlm.nih.gov/21963792/). Most recently, I have determined the first crystal structures of TDP1 in complex with small molecule inhibitors in collaboration with Dr. Yves Pommier and Dr. Terrence Burke. Current efforts are focused on developing new inhibitors of TDP1 based on our structural studies (https://pubmed.ncbi.nlm.nih.gov/34163656/). I am also collaborating with Dr. Maria Miller (Protein Structure Section, Wlodawer Lab) to determine the structural basis for interactions of C/EBPβ with transcriptional coactivators.
Biocatalysis:
In collaboration with Professor Lesley-Ann Giddings (Department of Chemistry, Smith College), we are studying the crystal structures and biochemistry of enzymes involved in the biosynthesis of siderophores and metal-chelating secondary metabolites that help microorganisms thrive in iron-limited environments. These enzymes are potentially useful as biocatalysts in the synthesis of clinically-approved siderophores and could be exploited as novel drug targets to combat certain human pathogens. I have determined the crystal structure of the cadaverine N-hydroxylase involved in desferrioxamine B biosynthesis in Streptomyces sviceus and its complexes with co-factors and natural substrates (https://pubmed.ncbi.nlm.nih.gov/33784308/).
Cryo-electron Microscopy:
The energy-dependent Lon proteases play a key role in cellular regulation by degrading short-lived regulatory proteins and misfolded proteins in the cell. Under the direction of Dr. Alexander Wlodawer, we are using single particle cryo-electron microscopy to study the full-length structure of the E. coli LonA protease. Recently, we determined the structure of the catalytically inactive S679A mutant of E. coli LonA protease by cryo-EM at 3.5 Å resolution without a bound substrate (https://pubmed.ncbi.nlm.nih.gov/34235464/). EcLonA without a bound substrate adopts a hexameric open-spiral quaternary structure that might represent the resting state of the enzyme that is poised to bind to substrate. Our ongoing research efforts are to solve structures of LonA protease bound to various substrates to study the dynamics of structural changes upon substrate binding and processing.
Publications
- Bibliography Link
- View Dr. Lountos' Full PubMed Summary.
- Google Scholar
Cryo-EM structure of substrate-free E. coli Lon protease provides insights into the dynamics of Lon machinery
Identification of a ligand binding hot spot and structural motifs replicating aspects of tyrosyl-DNA phosphodiesterase I (TDP1) phosphoryl recognition by crystallographic fragment cocktail screening
Structural characterization of inhibitor complexes with checkpoint kinase 2 (Chk2), a drug target for cancer therapy
Utilization of nitrophenylphosphates and oxime-based ligation for development of nanomolar affinity inhibitors of the Yersinia pestis outer protein H (YopH) phosphatase
Cellular inhibition of checkpoint kinase 2 (Chk2) and potentiation of camptothecins and radiation by the novel Chk2 inhibitor PV1019
Biography

George T. Lountos, Ph.D.
Dr. Lountos received his undergraduate degree in chemistry with high honor (2000) from the Georgia Institute of Technology where he performed research in organic chemistry in the laboratory of Prof. Suzy Beckham. He obtained a Ph.D. in chemistry (2005) from the School of Chemistry & Biochemistry, Georgia Institute of Technology, under the direction of Prof. Allen Orville where he conducted crystallographic studies of flavin-dependent enzymes. During his graduate studies, he was a recipient of the Georgia Tech Outstanding Graduate Teaching Assistant Award. From 2006-2011, he was a postdoctoral fellow in the Protein Engineering Section of the Macromolecular Crystallography Laboratory, under the direction of Dr. David Waugh. His postdoctoral research focused on structural studies of proteins involved in the Yersinia pestis type III secretion system and structure-based drug design of inhibitors of the human checkpoint kinase 2 and Yersinia pestis YopH. As a postdoc, Dr. Lountos was actively involved in the CCR Fellows & Young Investigators Committee and was appointed as the NCI-Frederick Postdoctoral Representative to the NIH Fellows Committee.
Dr. Lountos was the recipient of a Federal Technology Transfer Award in 2008 and the NIH Fellows Award for Research Excellence (FARE) in 2009 and in 2010. In 2011, he was hired as Scientist I with the Basic Research Program, Leidos Biomedical Research, Inc., where he works in support of the Macromolecular Crystallography Laboratory. In 2016, he was a recipient of the Special Achievement Award during the 19th Annual Frederick National Laboratory Achievement Awards Program. Dr. Lountos is an active member of the American Crystallographic Association (ACA) where he served as chair of the ACA Young Scientist Scientific Interest Group (2015). In 2017, he was appointed to the ACA Council where he served as the Young Scientist Representative to council until January 2020. He has been very active in the ACA National Meetings where he has organized and chaired several scientific sessions including the 2020 National Meeting Plenary Session on "Structural Contributions to SARS-CoV2 and the COVID-19 Pandemic". Dr. Lountos was elected to serve as Chair of the ACA Biological Molecules Scientific Interests Group in 2024. In 2020, Dr. Lountos was selected as a participant in the Cold Spring Harbor Laboratory Cryo-electron Microscopy Course. Dr. Lountos currently serves on the NCI Frederick Scientific Library Users Committee and has served as a research mentor in the NIH Summer Internship Program in Biomedical Research.
Covers

Identification of a ligand binding hot spot and structural motifs replicating aspects of tyrosyl-DNA phosphodiesterase I (TDP1) phosphoryl recognition by crystallographic fragment cocktail screening
Tyrosyl DNA-phosphodiesterase I (TDP1) repairs type IB topoisomerase (TOP1) cleavage complexes generated by TOP1 inhibitors commonly used as anticancer agents. TDP1 also removes DNA 3′ end blocking lesions generated by chain-terminating nucleosides and alkylating agents, and base oxidation both in the nuclear and mitochondrial genomes. Combination therapy with TDP1 inhibitors is proposed to synergize with topoisomerase targeting drugs to enhance selectivity against cancer cells exhibiting deficiencies in parallel DNA repair pathways. A crystallographic fragment screening campaign against the catalytic domain of TDP1 was conducted to identify new lead compounds. Crystal structures revealed two fragments that bind to the TDP1 active site and exhibit inhibitory activity against TDP1. These fragments occupy a similar position in the TDP1 active site as seen in prior crystal structures of TDP1 with bound vanadate, a transition state mimic. Using structural insights into fragment binding, several fragment derivatives have been prepared and evaluated in biochemical assays. These results demonstrate that fragment-based methods can be a highly feasible approach toward the discovery of small-molecule chemical scaffolds to target TDP1, and for the first time, we provide co-crystal structures of small molecule inhibitors bound to TDP1, which could serve for the rational development of medicinal TDP1 inhibitors.
See: Identification of a ligand binding hot spot and structural motifs replicating aspects of tyrosyl-DNA phosphodiesterase I (TDP1) phosphoryl recognition by crystallographic fragment cocktail screening by George T. Lountos, Xue Zhi Zhao, Evgeny Kiselev, Joseph E. Tropea, Danielle Needle, Yves Pommier, Terrence R. Burke, Jr. and David S. Waugh in Nucleic Acids Research, 2019, 47, 10134–10150.
Biomolecular Interactions of small-molecule inhibitors affecting the YopH protein tyrosine phosphatase
We have developed competitive and direct binding methods to examine small-molecule inhibitors of protein tyrosine phosphatase activity. Focusing on the Yersinia pestis outer protein H, a potent bacterial protein tyrosine phosphatase, we describe how an understanding of the kinetic interactions involving Yersinia pestis outer protein H, peptide substrates, and small-molecule inhibitors of protein tyrosine phosphatase activity can be beneficial for inhibitor screening, and we further translate these results into a microarray assay for high-throughput screening
Hogan, M., Bahta, M., Cherry, S., Lountos, G.T., Tropea, J.E., Zhao, B., Burke Jr., T.R., Waugh, D.S., and Ulrich, R.G. (2013). Biomolecular interactions of small-molecule inhibitors affecting the YopH protein tyrosine phosphatase. Chem. Biol. Drug Des. 81:323-333
Role of Glu312 in binding and positioning of the substrate for the hydride transfer reaction in choline oxidase
The crystal structure of choline oxidase provides mechanistic insights and reveals a novel FAD C4a-adduct. The solvent accessible surfaces of the homodimeric enzyme are colored to illustrate the dimer interface and the major domains within one subunit. The FAD binding (blue) and substrate binding (green) domains are partially separated by a loop (orange) that sequesters the active site within the interior of the enzyme
*Quaye, O., *Lountos, G.T., Fan, F., Orville, A.M., and Gadda, G. (2008). Role of Glu312 in binding and positioning of the substrate for the hydride transfer reaction in choline oxidase. Biochemistry 47: 243-256. * authors contributed equally
Small molecule microarray identifies inhibitors of tyrosyl-DNA phosphodiesterase 1 that simultaneously access the catalytic pocket and two substrate binding sites
Tyrosyl-DNA phosphodiesterase 1 (TDP1) is a member of the phospholipase D family of enzymes, which catalyzes the removal of both 3′- and 5′-DNA phosphodiester adducts. Importantly, it is capable of reducing the anticancer effects of type I topoisomerase (TOP1) inhibitors by repairing the stalled covalent complexes of TOP1 with DNA. It achieves this by promoting the hydrolysis of the phosphodiester bond between the Y723 residue of human TOP1 and the 3′-phosphate of its DNA substrate. Blocking TDP1 function is an attractive means of enhancing the efficacy of TOP1 inhibitors and overcoming drug resistance. Previously, we reported the use of an X-ray crystallographic screen of more than 600 fragments to identify small molecule variations on phthalic acid and hydroxyquinoline motifs that bind within the TDP1 catalytic pocket. Yet, the majority of these compounds showed limited (millimolar) TDP1 inhibitory potencies. We now report examining a 21 000-member library of drug-like Small Molecules in Microarray (SMM) format for their ability to bind Alexa Fluor 647 (AF647)-labeled TDP1. The screen identified structurally similar N,2-diphenylimidazo[1,2-a]pyrazin-3-amines as TDP1 binders and catalytic inhibitors. We then explored the core heterocycle skeleton using one-pot Groebke–Blackburn–Bienayme multicomponent reactions and arrived at analogs having higher inhibitory potencies. Solving TDP1 co-crystal structures of a subset of compounds showed their binding at the TDP1 catalytic site, while mimicking substrate interactions. Although our original fragment screen differed significantly from the current microarray protocol, both methods identified ligand–protein interactions containing highly similar elements. Importantly inhibitors identified through the SMM approach show competitive inhibition against TDP1 and access the catalytic phosphate-binding pocket, while simultaneously providing extensions into both the substrate DNA and peptide-binding channels. As such, they represent a platform for further elaboration of trivalent ligands, that could serve as a new genre of potent TDP1 inhibitors.
Zhao, X. Z.; Kiselev, E.; Lountos, G. T.; Wang, W.; Tropea, J. E.; Needle, D.; Hilimire, T. A.; Schneekloth, J. S.; Waugh, D. S.; Pommier, Y.; Burke, T. R. Small molecule microarray identifies inhibitors of tyrosyl-DNA phosphodiesterase 1 that simultaneously access the catalytic pocket and two substrate binding sites. Chem. Sci. 2021, 12, 3876, DOI: 10.1039/D0SC05411A

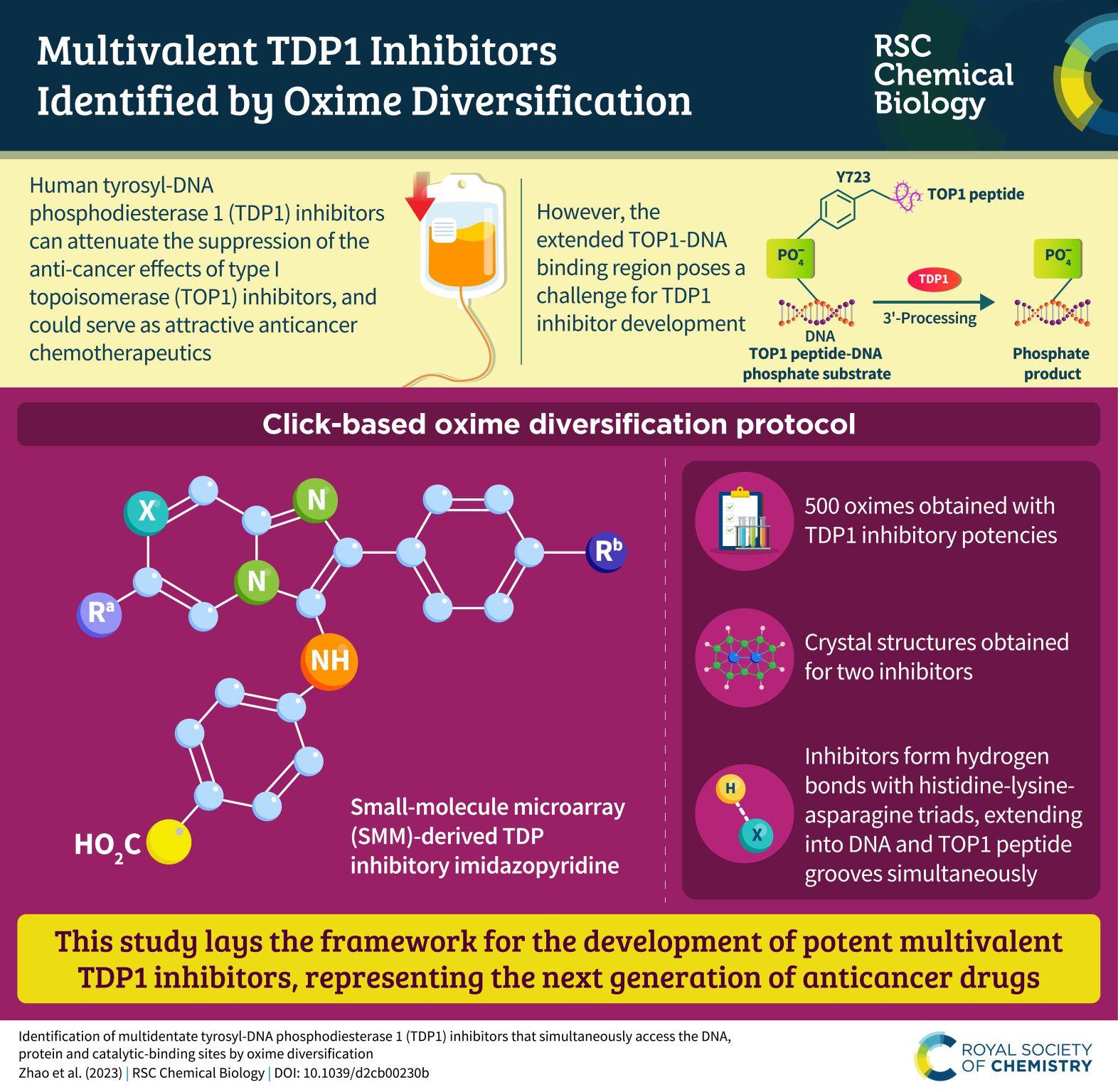
Identification of multidentate tyrosyl-DNA phosphodiesterase 1 (TDP1) inhibitors that simultaneously access the DNA, protein and catalytic-binding sites by oxime diversification
Tyrosyl-DNA phosphodiesterase 1 (TDP1) is a member of the phospholipase D family that can downregulate the anticancer effects of the type I topoisomerase (TOP1) inhibitors by hydrolyzing the 3'-phosphodiester bond between DNA and the TOP1 residue Y723 in the critical stalled intermediate that is the foundation of TOP1 inhibitor mechanism of action. Thus, TDP1 antagonists are attractive as potential enhancers of TOP1 inhibitors. However, the open and extended nature of the TOP1-DNA substrate-binding region has made the development of TDP1 inhibitors extremely challenging. In this study, starting from our recently identified small molecule microarray (SMM)-derived TDP1-inhibitory imidazopyridine motif, we employed a click-based oxime protocol to extend the parent platform into the DNA and TOP1 peptide substrate-binding channels. We applied one-pot Groebke-Blackburn-Bienayme multicomponent reactions (GBBRs) to prepare the needed aminooxy-containing substrates. By reacting these precursors with approximately 250 aldehydes in microtiter format, we screened a library of nearly 500 oximes for their TDP1 inhibitory potencies using an in vitro florescence-based catalytic assay. Select hits were structurally explored as their triazole- and ether-based isosteres. We obtained crystal structures of two of the resulting inhibitors bound to the TDP1 catalytic domain. The structures reveal that the inhibitors form hydrogen bonds with the catalytic His-Lys-Asn triads ("HKN" motifs: H263, K265, N283 and H493, K495, N516), while simultaneously extending into both the substrate DNA and TOP1 peptide-binding grooves. This work provides a structural model for developing multivalent TDP1 inhibitors capable of binding in a tridentate fashion with a central component situated within the catalytic pocket and extensions that project into both the DNA and TOP1 peptide substrate-binding regions.
Xue Zhi Zhao, Wenjie Wang, George T. Lountos, Evgeny Kiselev, Joseph E. Tropea, Danielle Needle, Yves Pommier and Terrence R. Burke, Jr.
Gallery
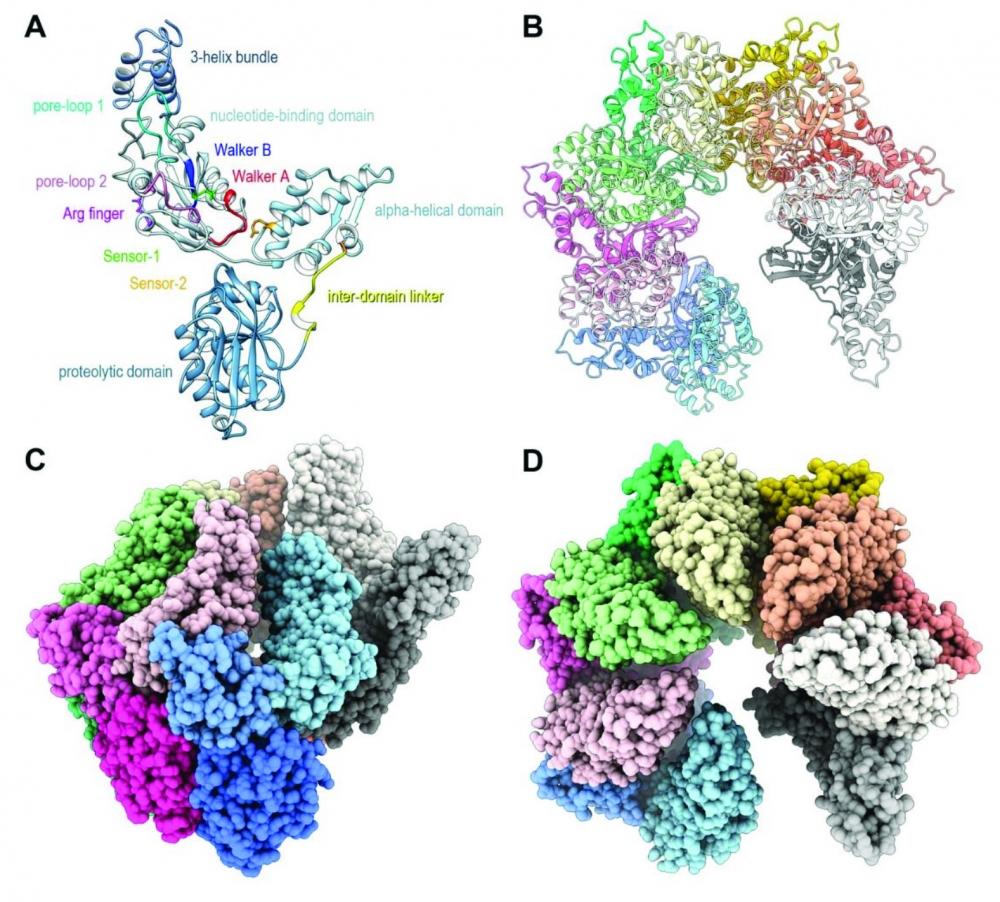
Cryo-EM structure of E. coli Lon A protease

2016 Frederick National Laboratory Special Achievement Award

2009 NIH Fellows Award for Research Excellence Award Ceremony, Bethesda, MD
Lab Life
https://ncifrederick.cancer.gov/about/theposter/content/magic-frederick-collaboration-yields-encouraging-results