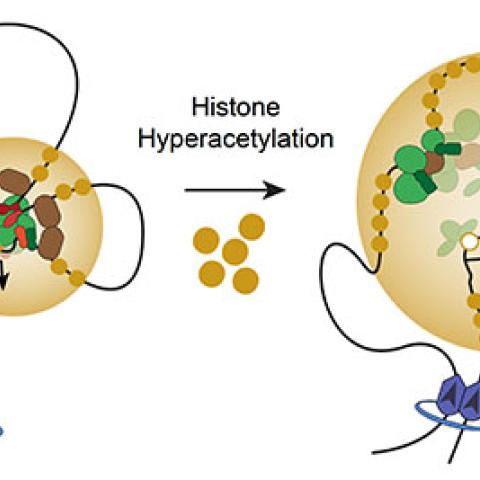
A model whereby histone hyperacetylation disrupts normal looping interactions, reduces binding of certain CR TFs, reduces binding of RNA Pol II, and causes loss of transcription at SE-dependent CR TF genes.
Image courtesy of Berkley Gryder
New CCR research suggests that a class of drugs known as histone deacetylase inhibitors can shut down a type of genetic circuit whose overactivity drives the growth of many cancers.
The discovery comes from studies of rhabdomyosarcoma, a cancer often caused by a mutation that alters the regulation of a gene network dedicated to overseeing muscle cell development. A research team led by Javed Khan, M.D., Deputy Chief of CCR’s Genetics Branch, has revealed an unexpected mechanism that usually helps keep this cell-identity circuit under tight control. The finding suggests a new way to treat not just rhabdomyosarcoma but also other cancers driven by the reprogramming of such circuits.
Rhabdomyosarcoma, which is most commonly diagnosed in children, develops in the body’s soft tissues, usually from immature muscle cells. In previous studies, Khan and postdoctoral research fellow Berkley Gryder, Ph.D., learned that rhabdomyosarcomas with a genetic rearrangement known as a PAX fusion have widespread disruptions to the chemical marks on the genome that influence how and when genes are turned on. These changes, which impact an interconnected network of regulators known as a core-regulatory circuit, impede muscle cell development and promote cell growth.
Because the genes that underlie cells’ core-regulatory circuits are regulated differently than other genes, Khan and Gryder suspected there might be a way to selectively intervene in this network, shutting it down to stop cancer cells’ growth.
Working with CCR’s Molecular Targets Laboratory, the researchers screened tens of thousands of compounds for their effects on rhabdomyosarcoma cells. They were seeking compounds that disabled the cells’ core-regulatory circuit without disturbing other genes. On July 8, 2019, Khan and Gryder, together with Dana-Farber Cancer Institute researcher Jun Qi, Ph.D., and other colleagues, reported in Nature Communications that the drugs that did this best were those that altered cells’ ability to process and interpret an epigenetic modification called histone acetylation.
It was no surprise that impeding histone acetylation or interfering with cells’ ability to read these marks dampens the activity of genes in core-regulatory circuits, Gryder says. Histone acetylation is usually associated with increased gene activity, and its marks are highly concentrated on the regulatory elements that control core-regulatory circuits. The discovery that target genes’ activity also slowed when cells were treated with compounds that allowed acetylation to build up, however, was puzzling. Those compounds, the histone deacetylase inhibitors, block the enzymes that remove acetylation marks from DNA-packing proteins, and were best known for their ability to turn genes on.
The team wondered whether the drugs’ effects might be due to an unusually complicated mechanism that keeps core-regulatory circuits under tight control. Unlike most genes, these networks must be activated by super-enhancers, a series of regulatory elements that lie far away from the genes they control. The super-enhancers’ targets only become active when a core transcription factor triggers the DNA to loop around and bring an enhancer element close to a gene’s start site. Khan and Gryder speculated that if too many acetyl groups crowded the regions between enhancer elements, the DNA might become less flexible and unable to make this connection.
To investigate histone acetylation’s impact on super-enhancers’ three-dimensional shape, the team turned to a method called HiChiP, which is used to map physical contact points within the genome. To definitively compare the genome’s architecture under different conditions, Khan, Gryder, and former NCI researcher Benjamin Stanton, Ph.D., developed a computational method called Absolute Quantification of Architecture (AQuA-HiChIP). Their method, currently in press at Nature Protocols, is being used by many research teams to investigate changes in genomic architecture.
Using AQuA-HiChIP, Khan, Gryder and colleagues showed that histone deacetylation inhibitors do indeed allow acetylation within super-enhancers to get out of control. “When you stop the eraser from working, the mark that you write starts to flood in every direction,” Gryder explains. Rather than clustering directly on enhancer elements, acetylation marks began to fill the spaces between them. As a result, the DNA fails to bend enough to activate the network. These findings, produced in collaboration with NIH’s National Institute of Biomedical Imaging and Bioengineering’s Advanced Imaging and Microscopy Resource and researchers at four other institutions around the world, were reported November 29, 2019, in Nature Genetics.
Khan says this new understanding gives researchers the information they need to begin exploring whether histone deacetylase inhibitors, likely in combination with other drugs, can treat rhabdomyosarcoma in patients who do not respond well to current therapies. “I think transcriptional poisoning is a very good avenue for therapy for this cancer,” he says. He’s also hopeful that the strategy will have a broader impact. Since core-regulatory circuits are all controlled by super-enhancers, histone deacetylase inhibitors may help shut down their activity in other cancers, too.