
Andre Nussenzweig, Ph.D.
- Center for Cancer Research
- National Cancer Institute
- Building 37, Room 1106A
- Bethesda, MD 20892-4254
- 240-760-7607
- 240-541-4489
- nussenza@mail.nih.gov
RESEARCH SUMMARY
Dr. Nussenzweig is a leading contributor to the study of mechanisms that maintain genomic stability and prevent cancer. His laboratory has elucidated many fundamental features of DNA damage and repair proteins and revealed the critical role they play in both normal and pathogenic states. Ongoing studies have emphasized the importance of DNA repair pathways as drivers of specific hematological malignancies and as contributors to chemoresistance/sensitivity in breast and ovarian cancers. The goal of his program is to use hypothesis-driven approaches to develop therapeutic strategies in the treatment of cancers.
Areas of Expertise
Andre Nussenzweig, Ph.D.
Research
Genomic instability is a hallmark of cancer. Central to a cell's ability to maintain genomic stability are systems that monitor and repair DNA double strand breaks (DSBs). DSBs occur during normal DNA replication, in response to chemotherapeutic agents, and during physiological reactions including meiotic recombination in germ cells and antigen receptor rearrangements in lymphocytes. If not rapidly and faithfully repaired, DSBs can also be substrates for aberrant chromosomal translocations, which promote cancer. The focus of the Recombination Unit is to understand the mechanisms by which all cell types monitor and repair DSBs
Resources
Protocol for END-seq
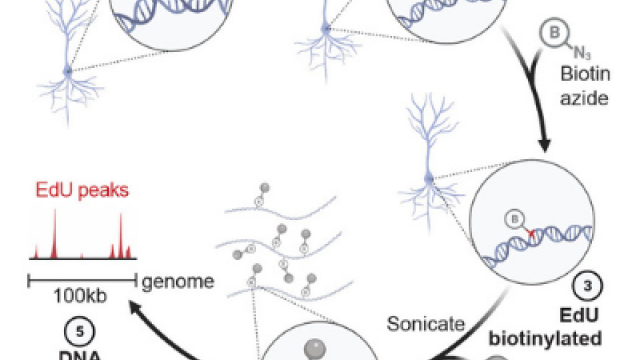
An unbiased and genome-wide approach that maps DNA double-strand breaks (DSBs) with precision and sensitivity. END-seq provides a snapshot of DNA ends genome-wide, which can be utilized for understanding genome-editing specificities and the influence of chromatin on a variety of physiological and pathological processes.

END-seq: An Unbiased, High-Resolution, and Genome-Wide Approach to Map DNA Double-Strand Breaks and Resection in Human Cells
Wong N, John S, Nussenzweig A, Canela A. Methods Mol Biol. 2021;2153:9-31
https://pubmed.ncbi.nlm.nih.gov/32840769/
DNA Breaks and End Resection Measured Genome-wide by End Sequencing
Canela A, Sridharan S, Sciascia N, Tubbs A, Meltzer P, Sleckman BP, Nussenzweig A. Mol Cell. 2016; 63:898-911
https://pubmed.ncbi.nlm.nih.gov/27477910/
Protocol for SAR-seq
Synthesis Associated with Repair sequencing - a method to map sites of DNA repair synthesis by sequencing.
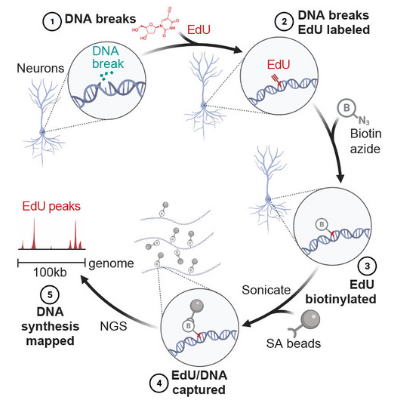
Neuronal enhancers are hotspots for DNA single-strand break repair
Wu, W, Hill, SE, Nathan, WJ, Paiano J, Callen E et. al. Nature. 2021; 593:440-444
https://pubmed.ncbi.nlm.nih.gov/33767446/
When the break is just around the loop
Genome Organization Drives Chromosome Fragility
Canela A, Maman Y, Jung S, Wong N, Callen E et. al. Cell. 2017;170:507-521
https://pubmed.ncbi.nlm.nih.gov/28735753
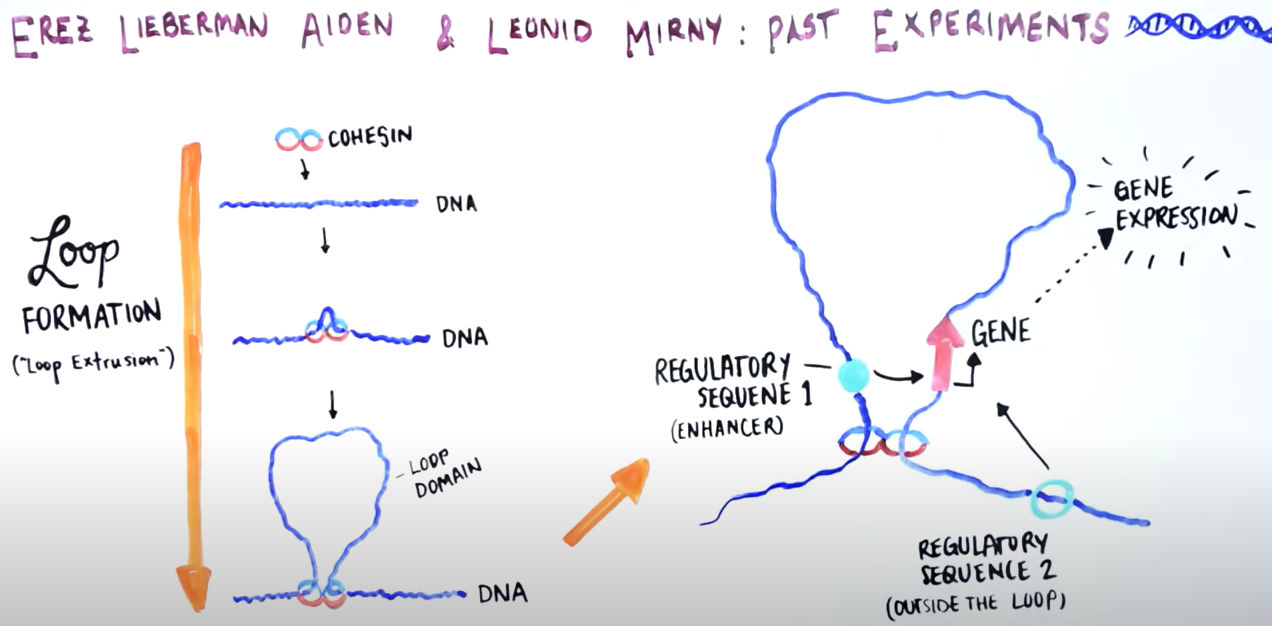
Inside the nucleus, DNA is packed in highly complex structures that help to control which genes are on and off each time. In this video abstract, we explain how this intricate architecture makes specific DNA sites more susceptible to breaks that, if not properly fixed, can lead to cancer.
Publications
Active DNA demethylation promotes cell fate specification and the DNA damage response
Neuronal enhancers are hotspots for DNA single-strand break repair
Repeat expansions confer WRN dependence in microsatellite-unstable cancers
Dual Roles of Poly(dA:dT) Tracts in Replication Initiation and Fork Collapse
Genome Organization Drives Chromosome Fragility
Biography

Andre Nussenzweig, Ph.D.
Dr. Nussenzweig received his Ph.D. in Physics from Yale University in 1989. He completed his postdoctoral training in atomic physics in Paris with Dr. Serge Haroche, who was awarded the Nobel prize in Physics in 2012. Subsequently, Dr. Nussenzweig became a Research Fellow at Memorial Sloan-Kettering Cancer Center prior to joining the Experimental Immunology Branch as a tenure track investigator in 1998. Dr. Nussenzweig received tenure at NIH in 2003. In 2011, Dr. Nussenzweig established a new department at NCI called the Laboratory of Genome Integrity. Dr. Nussenzweig is an elected member of the European Molecular Biology Organization, a National Institutes of Health Distinguished Investigator and a 2019 inductee into the US National Academy of Medicine. In 2023, Dr. Nussenzweig was inducted into the American Academy of Arts and Sciences as well as the National Academy of Sciences.
Job Vacancies
| Position | Degree Required | Contact Name | Contact Email |
|---|---|---|---|
| Postdoctoral Fellow - Computational biology, genomics | Ph.D. or equivalent | Sam John | sam.john@nih.gov |
| Postdoctoral Fellow - DNA damage, DNA repair | Ph.D. or equivalent | Sam John | sam.john@nih.gov |
Team















News







Alumni

Lab Life

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

LGI Summer Picnic - July 2021 - Lake Needwood

2019 - The Nussenzweig Lab

LGI Summer Picnic - July 2017 - Bethesda

LGI Summer Picnic - July 2017 - Bethesda

LGI Summer Picnic - July 2017 - Bethesda

LGI Summer Picnic - July 2017 - Bethesda

LGI Summer Picnic - July 2017 - Bethesda

LGI Summer Picnic - July 2017 - Bethesda

LGI Summer Picnic - July 2017 - Bethesda

LGI Summer Picnic - July 2017 - Bethesda

LGI Summer Picnic - July 2017 - Bethesda

LGI Summer Picnic - July 2017 - Bethesda
LGI Summer Picnic - July 2017 - Bethesda

LGI Summer Picnic - July 2017 - Bethesda

LGI Christmas Party - December 2016 - Pacifica Cafe, Gaithersburg

LGI Christmas Party - December 2016 - Pacifica Cafe, Gaithersburg

LGI Christmas Party - December 2016 - Pacifica Cafe, Gaithersburg

LGI Christmas Party - December 2016 - Pacifica Cafe, Gaithersburg

LGI Christmas Party - December 2016 - Pacifica Cafe, Gaithersburg
LGI Christmas Party - December 2016 - Pacifica Cafe, Gaithersburg

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood
LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood

LGI Summer Picnic - July 2015 - Lake Needwood